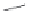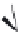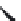Biomedical Engineering Reference
In-Depth Information
has resulted in
16.63
, which displays a signii cant functional preference as a potent agonist of a
4
b
2
and muscle-type nAChRs over other nAChRs. In contrast, (
)-UB-165 (
16.64
), a hybrid compound
between epibatidine and anatoxin-a (
16.56
, isolated from the alga
Anabaena l os aquae
), is one of
the few nAChR agonists available that preferentially activate the “minor” heteromeric nAChRs
without concomitant activation of the major CNS subtypes.
(−)-Cytisine (
16.54
) from
Laburnum anagyroides
is a potent partial agonist of the heteromeric
nAChRs. (−)-Cytisine and its analog varenicline (
16.65
) have recently been launched as smoking
cessation aids. Further, the complex natural source compound lobeline (
16.57
) from
Lobelia inl ata
is also under clinical development for treatment of smoking dependence.
Although the homomeric a
7
nAChR represents a low-afi nity binding site for ACh and the natural
source compounds
16.52 -16.56
, it has been possible to develop potent and a
7
selective agonists from
some of these leads. The toxin anabaseine (
16.55
) is isolated from marine worms and certain ant spe-
cies, and it is a rather nonselective agonist displaying a somewhat higher efi cacy at a
7
nAChR than at
the heteromeric nAChRs. Introduction of conjugated aryl substituents in the 3-position of the pyridine
ring of anabaseine has increased this selectivity, as exemplii ed by the prototypic a
7
agonist GTS-21
(
16.66
). The quinuclidine (1-azabicyclo[2.2.2]octane) ring system forms the scaffold in several
a
7
-selective agonists, including AR-R-17779 (
16.67
) and PNU-282987 (
16.68
), of which the latter has
entered clinical trials for schizophrenia. Finally, replacement of the pyrrolidine ring in
16.52
with a
azabicyclo[3.2.2.]nonane ring has provided the potent and selective a
7
agonist TC-1698 (
16.69
).
±
16.5.2 nAChR A
NTAGONISTS
As described for the nAChR agonists, several competitive nAChR antagonists have been obtained
from natural sources. The peptide toxin, a-bungarotoxin (
16.70
) from the Taiwan banded krait
(
Bungarus multicinctus
) is a potent competitive antagonist of a
7
and muscle-type nAChRs, and
methyllycaconitine (
16.71
), isolated from
Delphinum
and
Consolida
species, is a highly selective a
7
antagonist. In contrast to the selectivity of these two compounds, other classical competitive nAChR
antagonists, including dihydro-b-erythroidine (DHbE,
16.72
), are far less discriminative between
different nAChR subtypes (Figure 16.14).
In addition to the agonists derived from (
S
)-nicotine and (
)-epibatidine, several antagonists have
emerged. Introduction of
n
-alkyl groups ranging from methyl to dodecyl (C
12
H
25
) at the pyridine
nitrogen of (
S
)-nicotine have produced several potent albeit nonselective antagonists (exemplii ed
by
16.73
). Furthermore, the pyridyl ether A-186253 (
16.74
), a
16.58
analog, displays high selectivity
±
O
CC
C
C
O
O
H
MII
PnIA
H
N
O
O
OH
O
O
N
O
N
H
OH
O
O
O
N
N
α-Bungarotoxin (
16.70
)
DHβE (
16.72
)
MLA (
16.71
)
ImI
AuIB
NH
2
Cl
Cl
N
N
O
N
N
N
+
Cl
N
(
16.73
)
A-186253 (
16.74
)
(
16.75
)
N
N
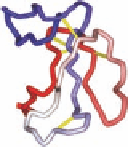
FIGURE 16.14
Chemical structures of competitive nAChR antagonists and 3D structures of a-bungarotoxin
and four a-conotoxins.