HDV is extremely prolific. The serum of an infected
There is now considerable evidence that all TSEs are
individual can contain up to 1012 RNA-containing HDV
related and result from defects in the metabolism of the prion
particles per milliliter.
protein. The pattern of symptoms associated with a particular
TSE may vary, however, depending in part on how the dis-
ease was contracted; on the source of the infecting agent;
Host Range of HDV
and on the nature of mutations in the prion protein. Thus,
The only known natural hosts for HDV are humans,
although the prion protein is central to disease in every case,
but HDV can be experimentally transmitted to chimpan-
symptomology can differ, in part because the particular area
zees and to woodchucks. Infection of chimps requires
of the brain most affected can vary.
coinfection with HBV, and this provides a useful primate
There is no immune response associated with any TSE.
model for the study of the agent. A second model system is
No antibodies are formed and no inflammation marked by
furnished by woodchucks. Woodchucks can be chronically
the infiltration of mononuclear cells is present. As stated, it
infected with woodchuck hepatitis virus (WHV, Chapter
is relentlessly progressive and always results in death.
6), a relative of HBV, and WHV can provide helper activ-
ity for HDV. Chronically infected animals can be infected
Kuru
with HDV, and in this case the surface properties of the
HDV virion are determined by the helper WHV rather than
Kuru was a disease of epidemic proportions among the
by HBV.
Fore people of New Guinea that reached a prevalence of
about 1% of the population. The disease was characterized by
progressive ataxia that led to total incapacitation and death,
PRIONS AND PRION DISEASES
normally in 1218 months after the appearance of symptoms
in adults or 312 months in children. The demonstration that
Transmissible spongiform encephalopathies (TSEs),
kuru was transmissible to primates by inoculation of brain
now often referred to as prion diseases, are progressive,
tissue from people dying of the disease was the first dem-
fatal diseases of humans and of other animals. A listing of
onstration of the transmissibility of a TSE in humans. These
TSEs is given in Table 9.2. TSEs of humans include kuru,
transmission studies and other studies of kuru resulted in a
Creutzfeldt-Jakob disease (CJD), Gerstmann-Straüssler-
Nobel prize for Carleton Gajdusek in 1976, the first of two
Scheinker syndrome (GSS), and fatal familial insomnia
prizes for work with TSEs (Chapter 1).
(FFI). TSEs are characterized by neuronal loss that appears
Kuru is believed to have been spread among the Fore
as a spongiform degeneration in sections of brain tissue,
people by cannibalism in which the bodies of relatives who
often accompanied by amyloid plaques or fibrils. The most
had died were eaten in a ritualistic feast. Women and chil-
prominent symptoms of disease are usually dementia (loss of
dren were more often affected than men, and it is thought
intellectual abilities) or ataxia (loss of muscle control during
this was because they prepared the body for the feast and
voluntary movement) that results from the progressive loss
they ate the brains of deceased relatives. Men were less often
of brain function. The disease always has a fatal outcome. In
affected, it is conjectured, because they ate primarily other
humans, death usually occurs within 6 months to 1 year of
body parts. It has been postulated that the epidemic began
the first appearance of symptoms.
when a member of the tribe died of a sporadic case of CJD,
TSEs can be contracted by inoculation with or inges-
and the disease was then spread to others through cannibal-
tion of brain tissue or other tissues containing the infectious
ism. Through the efforts of missionaries, cannibalism ceased
agent, and thus they can be transmitted as an infectious
many years ago and the disease has become progressively
disease. Kuru first came to light as an infectious disease and
rarer. Now only older people who contracted the infectious
many cases of CJD in humans have been acquired by infec-
agent during the time of cannibalism continue to develop the
tion. However, TSEs can also occur as sporadic diseases for
illness. From studies of the continuing development of kuru
which there is no evidence of infection by an outside agent.
in older Fore people, it is known that the disease can appear
In humans, CJD occurs sporadically with a frequency of
as long as 40 years after the event that resulted in infection
about 10-6. Finally, TSEs can appear as inherited diseases.
with the agent.
GSS, most FFI, and some cases of CJD occur as dominant
inherited diseases, associated with mutations in the gene for
Sporadic and Iatrogenic CJD
the prion protein. Inheritance of the mutant gene dramati-
cally increases the probability of developing TSE, such that
CJD in man is usually a sporadic illness that occurs with
a frequency of about 10-6 that is uniform around the world.
the probability of acquiring the disease over a lifetime may
approach 100%. In most cases of sporadic or inherited TSE,
However, once the disease has arisen it is transmissible by
the disease is transmissible as an infectious disease once it
inoculation of infected material into experimental animals
occurs.
such as primates and transgenic mice. CJD has also been
from unequal crossing over, in some cases of inherited CJD
transmitted iatrogenically to humans. Iatrogenic cases have
or GSS. These three diseases are distinguished on the basis
occurred in recipients of pituitary-derived human growth
of symptomology, which is overlapping. CJD is character-
hormone obtained from cadavers, some of whom died of
ized by ataxia, dementia, and behavioral disturbances. GSS
CJD; in recipients of homographs of dura mater derived
is usually characterized by cerebellar disorders accompa-
from cadavers; through implantation into epilepsy patients
nied by a decline in cognitive ability. FFI, as its name sug-
of contaminated silver electrodes that had been incompletely
gests, is characterized by abnormal sleep patterns, including
sterilized; and through corneal transplants. The infec-
intractable insomnia.
tious agents of TSEs are extremely difficult to inactivate
The penetrance of the different mutations varies but is
and require extraordinary sterilization techniques in order
usually very high. For example, CJD caused by the change
to destroy their infectivity. Better methods of sterilization
from glutamic acid-200 to lysine (E200K), when residue 129
have been introduced, and human growth hormone is now
is homozygous for methionine, has been estimated to have a
produced in bacteria from recombinant DNA plasmids, so
penetrance of 0.45 by age 60 and a penetrance of more than
that the iatrogenic spread of CJD has been greatly reduced.
0.96 above age 80. Thus, a person with this mutation is almost
Sporadic FFI has also been described. No case of sporadic
certain to develop CJD if he or she lives long enough.
GSS is known, however.
Attempts have been made in many cases of inherited
TSEs to transmit the disease to subhuman primates or to
Inherited For ms of Human TSE
mice. Transmission has been achieved in most cases tested.
About 5% of CJD cases arise in a familial, autosomal
Thus once the disease arises, it is transmissible to animals
dominant fashion and are associated with mutations in the
that do not contain the mutation.
gene for the prion protein. GSS and most cases of FFI are
In addition to mutations associated with inherited TSEs,
also inherited forms of TSE associated with mutations in
several polymorphisms in the prion protein are known that are
the prion protein. Many of the responsible mutations are
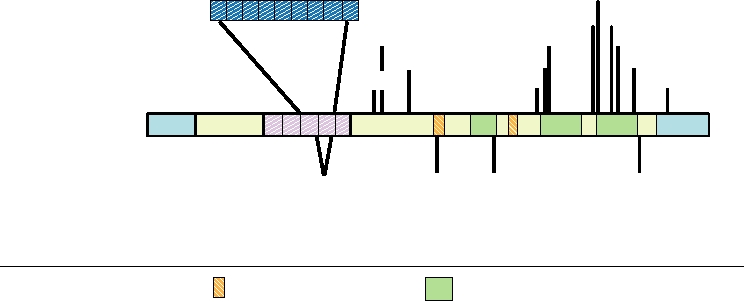
not associated with disease (Fig. 9.10). The polymorphism
illustrated in Fig. 9.10, in which a schematic diagram of the
at residue 129 is of particular importance. Homozygosity at
human prion protein is presented.
this position affects the probability of contracting TSE.
A dozen single amino acid substitutions in the prion pro-
tein have been found to be associated with inherited CJD,
TSEs in Other Animals
GSS, or FFI. Additionally, an element normally contain-
ing five repeats of a 24-nucleotide sequence (encoding an
Naturally occurring TSEs of a number of other mammals
8-amino-acid repeat, P-Q/H-G-G-G-W-C-Q) has been found
are known. The oldest known TSE, in fact, is that of sheep, and
to contain one to nine extra repeats, probably originating
is called scrapie. Scrapie has been known for more than 200
E200K
F198S R208H
Polymorphisms associated
P105L
T183A
V210I
Insertion of
with prion disease
V180I
Q217R
A117V
2-9 octarepeats
M232R
D178N
P102L
Pre HPrPc
H1
H2
H3
S
S
Polymorphisms that are
Deletion of an
M129V N171S
E219K
phenotypically wild type
octarepeat
Beta sheets
Alpha helices
H1
D178N- Point mutation associated with FFI
P102L - Point mutations associated with GSS
E200K- Point mutations and insertions associated with familial CJD
M129V - homozygosity at this locus increases susceptibility to sporadic CJD
FIGURE 9.10
Mutations found in the human prion protein gene. Polymorphisms that are phenotypically wild type are
shown below the schematic of the gene; mutations that segregate with inherited prion diseases are shown above the gene.
GSS, FFI, and CJD are defined in Table 9.3. Adapted from Prusiner (1998) and Riek et al. (1996).
TABLE 9.3 Prion Diseases
Disease (abbreviation)
Natural host
Experimental hosts
Cause of disease
Scrapie
Sheep and goats
Mice, hamsters, rats
Infection in genetically susceptible sheep
Transmissible mink encephalopathy (TME)
Mink
Hamsters, ferrets
Infection with prions from sheep or cattle
Chronic wasting disease
Mule deer, white
Ferrets, mice
Unknown
tail deer, and elk
Bovine spongiform encephalopathy (BSE)
Cattle
Mice
Infection with prion-contaminated meat and
bonemeal
Feline spongiform encephalopathy (FSE)
Cats
Mice
Infection with prion-contaminated beef
Exotic ungulate encephalopathy (EUE)
Nyala, oryx, and
Mice
Infection with prion-contaminated meat and
greater kudu
bonemeal
Kuru
Humans
Primates, mice
Infection through ritual cannibalism
Creutzfeldt-Jakob disease
Humans
Primates, mice
iCJD (iatrogenic)
Humans
Infection from prion-contaminated human
growth hormone, dura mater grafts, etc.
sCJD (sporadic)
Humans
Somatic mutation or spontaneous conversion of
PrPc to PrPSc
nvCJD (new variant)
Humans
Ingestion of bovine prions?
fCJD (familial)
Humans
Germ line mutation in PrP gene
Gerstmann-Straüssler-Scheinker
Humans
Germ line mutation in PrP gene
syndrome (GSS)
Fatal familia insomnia (FFI)
Humans
Primates, mice
Germ line mutation in PrP gene (D178N, M129)
Fatal sporadic insomnia (FSI)
Humans
Somatic mutation or spontaneous conversion of
PrPc to PrPSc
Source: Adapted from Granoff and Webster (1999), p. 1389.
years and is widely distributed in Europe, Asia, and America.
in Britain. The epidemic was maintained by feeding to cattle
The name comes from the tendency of animals to rub them-
the processed offal from cattle and other animals, that is, by a
selves against upright posts, apparently because of intense
form of animal cannibalism as happened with kuru in humans.
itching that arises from this neurological disease. Scrapie
Although this practice was of long standing, it did not cause
appears to be transmitted horizontally in sheep flocks, but the
trouble until recently, when a change in the rendering proc-
mechanism by which it is transmitted is not understood. The
ess was introduced. It is believed that this change allowed the
infectious agent is very resistant to inactivation and may per-
BSE agent to survive the processing steps, whereas formerly it
sist in pastures for a long time. It may be ingested, but other
had been killed during rendering. The result was an epidemic
mechanisms for persistence have also been proposed.
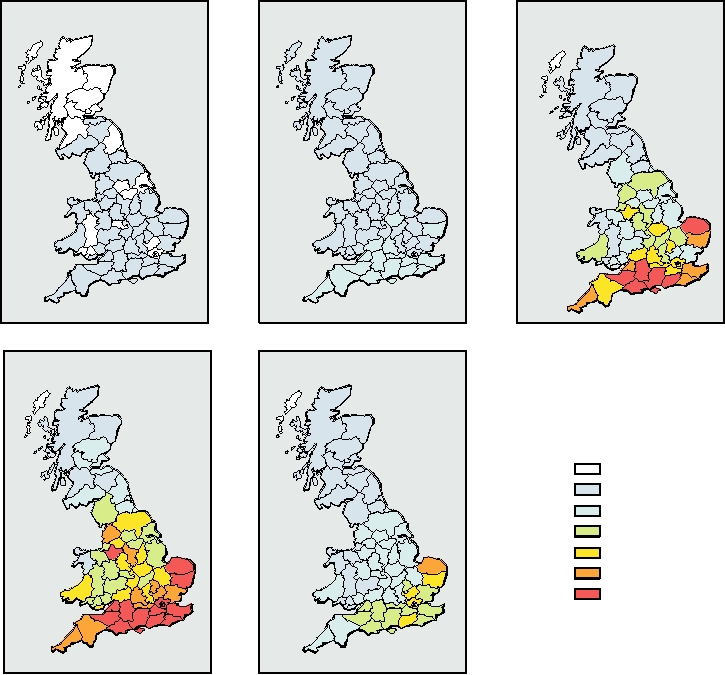
of BSE that spread across all of Britain (Fig. 9.11). At the
Scrapie appears to have been transmitted to a number of
height of the epidemic, there were more than 35,000 cases of
other mammals. In some cases the spread has been to animals
BSE per year in Britain (Fig. 9.12).
that share pasturage with infected sheep, such as white-tailed
The original source of the BSE that led to the epidemic
deer, mule deer, and elk (where the disease is called chronic
is uncertain. It may have arisen from a spontaneous case
wasting disease). In these cases, it is thought that infection
of BSE, similar to spontaneous CJD in humans, although
occurs by the same mechanisms that maintain scrapie in
spontaneous BSE in cattle appears to be rare or nonexistent.
sheep flocks. In other cases, spread has occurred via food
A second possibility is that it may have arisen from infec-
derived from infected sheep that was fed to mink (transmis-
tion with scrapie from infected sheep, since sheep offal was
sible mink encephalopathy), domestic cats or exotic cats in
included in the rendered offal.
zoos (feline spongiform encephalopathy), ungulates in zoos
Once the epidemic of BSE in cattle in Britain was recog-
(exotic ungulate encephalopathy), and perhaps to cattle.
nized, legislation was introduced that banned the feeding of
However, there is no evidence that scrapie has ever spread
any ruminant-derived protein to ruminants. Also introduced
to humans, despite the long history of human consumption
was legislation to make BSE a notifiable disease and to pro-
of scrapie-infected sheep.
hibit the use of brain, spinal cord, and certain other offals
Bovine spongiform encephalopathy (BSE), also called mad
from any bovine animal in human food. These initial bans
cow disease, is a TSE of cattle that was recently an epidemic
were subsequently enlarged and extended in various ways
1991
1987
1989
Avo
Avo
n
n
Avo
n
1993
1995
Incidence of BSE
Cases per 1000 head of cattle
None
<1
1 to 2
2 to 3
3 to 4
4 to 5
>5
Avo
Avo
n
n
FIGURE 9.11 Spread of the BSE epidemic in the British Isles. Geographic distribution of the incidence of BSE per
head of cattle by county from 1989 to 1995. Adapted from Anderson et al. (1996).
(see Fig. 9.12 and its legend). The ruminant feed ban resulted
occur in experimental systems, there is a requirement for
in the waning of the epidemic in cattle in Britain, but new
an adaptation event before the agent can be readily trans-
cases continued to arise, whether the result of a long latent
mitted. Humans were thought not to be sensitive to animal
period of the infectious agent, or from contaminated rumi-
TSE agents because of this species barrier. In particular, no
nant feed that continued to enter the system, or from alter-
evidence for the transmission of scrapie to humans has ever
native modes of transmission, such as passing the infection
been found despite the fact that people all over the world,
from mother to calf. With the recognition of new variant CJD
but especially in Britain, have eaten sheep infected with
in people, the issue of eradicating BSE became more pressing
scrapie for 2 centuries.
and culling of cattle was undertaken. This culling of herds
In 1995 and 1996, however, 12 cases of a variant form
containing BSE-infected cattle together with the subsequent
of human CJD occurred in Britain. These new variant CJD
culling of herds infected with foot-and-mouth disease virus,
cases (nvCJD) were characterized by an unusually early
as well as the continued enforcement of the ruminant feed
age of onset, with some cases in their teens, and by a dif-
bans, have resulted in a marked reduction in the incidence of
ferent symptomology. A comparison of the ages at which
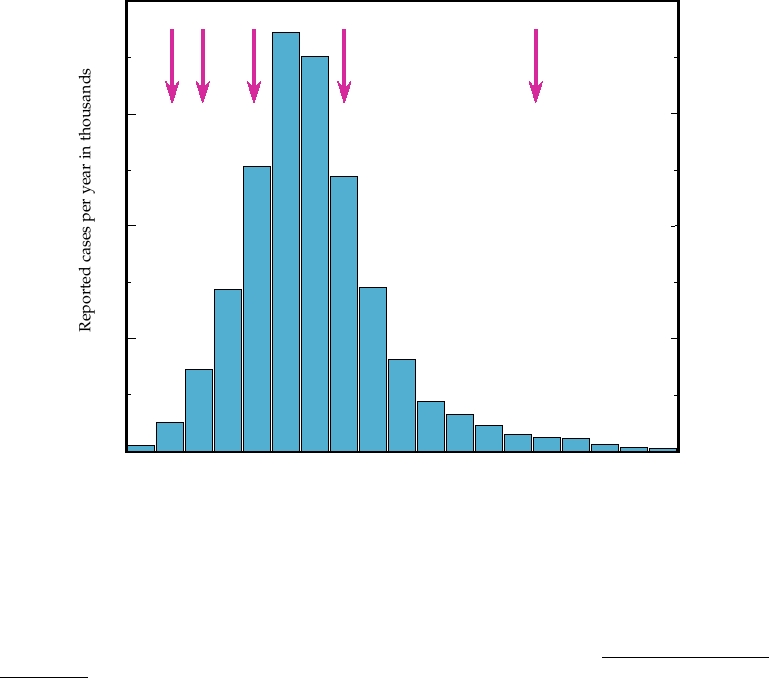
BSE, although not to its total eradication (Fig. 9.12).
people in Britain contracted sporadic CJD during the
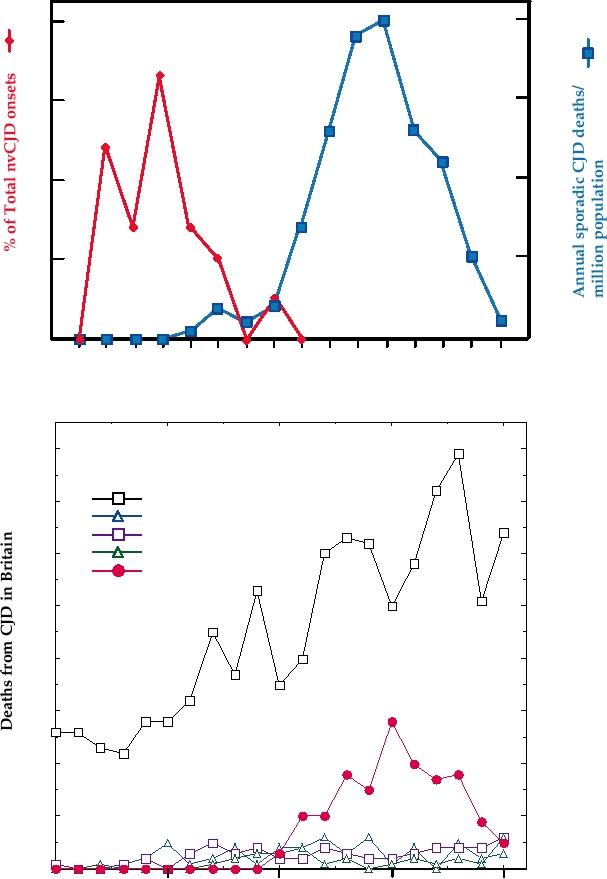
last 25 years with that of the ages of the first 21 cases of
nvCJD is shown in Fig. 9.13A. Sporadic CJD is primarily
New Variant CJD in Humans
a disease of people in their 50s, 60s, and 70s, with a peak
At the beginning of the BSE epidemic, public health
of occurrence in the early 60s. Cases in people under 40
officials in Britain had little fear that the epidemic might
are rare. Variant CJD to date has been a disease of young
pose a threat to human health. There is a species barrier
people, primarily people in their teens, 20s, and early 30s.
to the transmission of the TSE from any particular animal
Symptomology also differs. Sporadic CJD is characterized
to another animal. Even in cases where transmission does
by dementia as an early symptom, whereas variant CJD
40
1
2
3
4
5
30
20
10
0
87 88 89 90 91 92 93 94 95 96 97 98 99 00 01 02 03 04 05
Year
FIGURE 9.12 Confirmed cases of BSE (bovine spongiform encephalopathy) in British cattle per year between 1987
and 2005. Arrows indicate (1) ruminant feed ban (1988); (2) specified offals ban, to prevent offals proteins from entering
the human food chain (1989); (3) extended specified offals ban [prohibiting feeding of offal proteins to pigs and poultry
(1991)]; and (4) offals ban futher extended to include offals from bovines < 6 months old (1994). (5) Prohibit the use
of any animal protein (excluding milk and fish meal) from feed for any farmed animal species (2001). Note that data
after 2002 could be biased by the large number of cattle slaughtered during the foot-and-mouth-disease virus (FMDV)
epidemic in 2001, which must have contained some infected animals. Adapted from Anderson et al. (1996), the "2004
Institute of Food Science and Technology Information Statement on BSE," and data from http://www.oie.int/fr/info/
fr_esbru.htm.
is characterized by psychiatric symptoms, usually depres-
earlier, kuru has a long latent period, with disease devel-
sion, and the patient is often first seen by a psychiatrist.
oping as long as 40 years after infection. The decline in the
Third, time to death averages somewhat longer in variant
incidence of nvCJD following control of BSE, however,
CJD than in sporadic CJD. The number of cases of nvCJD
suggests that the dynamics of nvCJD disease are different
rose for several years, plateaued in the year 2000, and then
and that only small numbers of disease will continue to
declined, as shown in Fig. 9.13B. Also shown in this figure
arise, perhaps the result of the species barrier that exists
for comparison are the number of cases of sporadic CJD
for the transmission of BSE to humans. Further, it is not
each year in Britain; the rise in the number of cases of
understood why the young are so much more sensitive
sporadic CJD reported over this time frame is probably
to nvCJD than are the old. A sensationalized and grip-
due to increased recognition of CJD disease, catalyzed in
ping account of kuru, CJD, and BSE is found in the book
part by the nvCJD epidemic. Through 2005 there had been
Deadly Feasts by Richard Rhodes.
a total of about 150 cases of nvCJD.
There is now a considerable body of evidence that
Prion Protein
nvCJD is caused by infection with BSE and results from
eating BSE-contaminated meat. For one, the BSE prion
The nature of the infectious agents responsible for
and the human nvCJD prion are closely related and differ
scrapie and other TSEs has been controversial, in part
from other CJD prions (see later). For another, the nvCJD
because the study of these agents has presented enor-
epidemic closely parallels the BSE epidemic with an 8-
mous technical difficulties. The kuru agent was shown
year lag. It appears, therefore, that the incubation period
to be transmissible to other primates many years ago,
of nvCJD, at least to date, averages about 8 years. It is not
but the incubation period is very long in these animals
clear how many cases may ultimately arise. As described
(more than 10 years in some cases) and they are expen-
A
2.0
40
1.5
30
1.0
20
0.5
10
10
20
30
40
50
60
70
80
Age in years
B
80
Sporadic
70
Iatrogenic
Familial
GSS
60
nvCJD
50
40
30
20
10
0
1985
1990
1995
2000
2005
Year
FIGURE 9.13
Creutzfeldt-Jakob disease (CJD) in Britain. (A) Age distribution of the first 21 cases of new variant
CJD (nvCJD) in 1995 and 1996, compared with the annual age-specific death rates for sporadic CJD (573 cases) in
Britain between 1970 and 1994. The scales have been chosen to optimize comparison of the age distributions. Adapted
from Nathanson (1999) Figure 7 on p. 449. (B) Cases of CJD of various etiologies and Gerstmann-Straüssler-Scheinker
syndrome (GSS) in Britain from 1985 to 2005. The rise in the number of cases of sporadic CJD reported is probably due
to increased recognition of the disease. Data are Monthly CJD Statistics, from the Department of Health of the United
Kingdom.
sive to maintain, which limited early progress in the
have been very useful because the genetic background
study of the molecular biology of the agents. The sub-
can be controlled. However, such studies remain slow
sequent discovery that many TSEs could be transmitted
and tedious because an infectivity assay often takes more
to mice and hamsters, in which the incubation period
than 1 year.
was much shorter, as short as 60 days in some instances,
Studies in mice and other animals, as well as the find-
speeded up progress. Transgenic mice, in particular,
ing that mutations in the prion protein are associated with
inherited TSEs in humans, have made clear that the prion
protein, abbreviated PrP, is intimately involved in the trans-
mission of TSE and in the disease process. The normal cel-
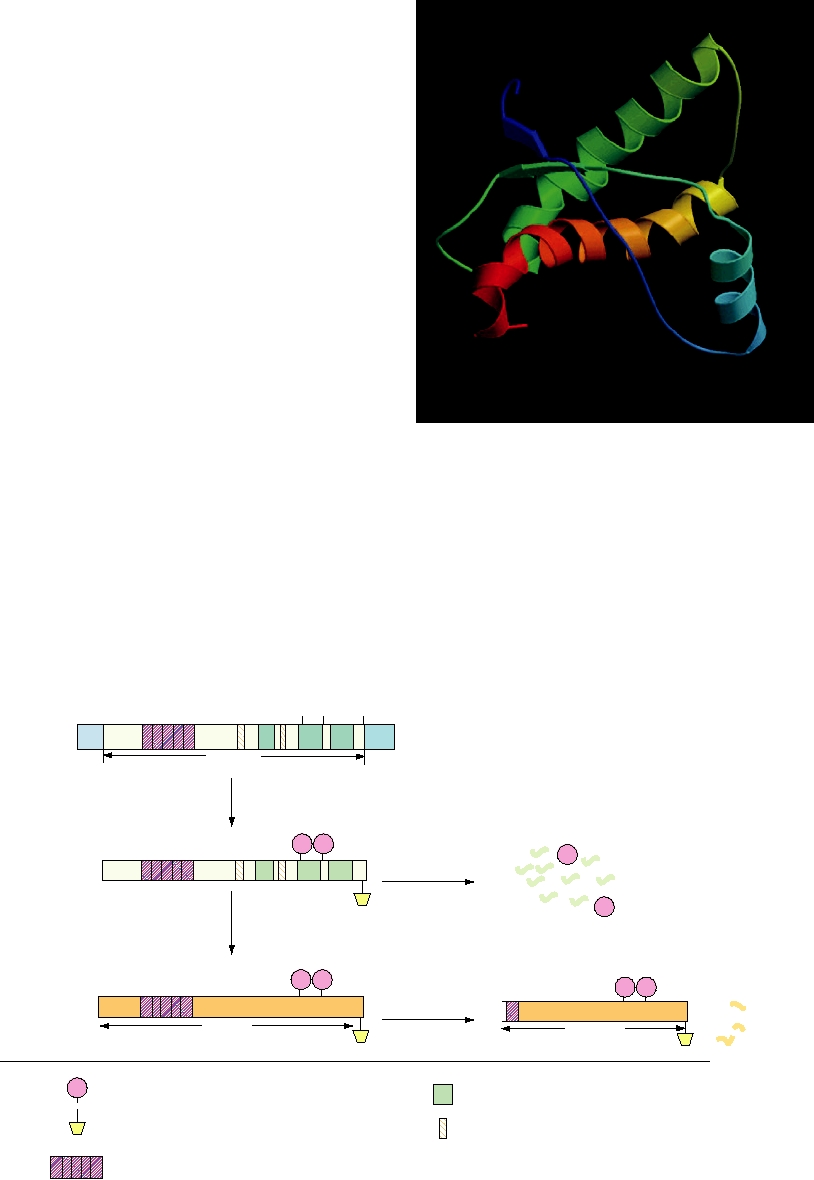
aa121
lular protein is referred to as PrPc. The structure in solution
of the C-terminal half of the mouse version of this protein
H2
S1
(residues 121231) is illustrated in Fig. 9.14. The protein has
a high content of α helix. In this half of the protein, there are
S2
three α-helical domains of 11, 15, and 18 residues, and only
a short (four residues in each strand) two-stranded antiparal-
lel β sheet. The N-terminal 98 residues of this protein form
a flexible random coil in solution, as determined by nuclear
H3
magnetic resonance imaging.
H1
The prion protein is synthesized as a larger precursor of
254 amino acids that contains both N-terminal and C-ter-
aa231
minal extensions (Fig. 9.15). The N-terminal extension is a
signal sequence that leads to the translocation of PrP into the
lumen of the endoplasmic reticulum. It is removed by signal
peptidase, as are most N-terminal signal sequences. The
C-terminal extension is removed by another cellular pro-
FIGURE 9.14 The structure of the prion protein. The structure of
tease and the protein is attached to a phosphoinositol gly-
residues 121231 of the mouse prion protein in solution as determined by
NMR is shown. The protein is color coded from blue at residue 121 to red
colipid anchor that anchors the protein in the membrane. The
at residue 231, with β sheets shown as flat arrows and α helices as coils.
protein is N-glycosylated on two sites. The processed pro-
The second and third helices are linked with a disulfide bond (not shown).
tein is transported to the plasma membrane and transiently
(Compare with Figures 9.10 and 9.15.) Adapted from Riek et al. (1996).
Precursor human prion protein PrPc
N N
S
181 197
231
H1
H2 H3
S
N
S
C
23 aa
22 aa
209aa
Maturation
Mature cellular prion protein PrPc
CHO CHO
CHO
Proteinase K
H1
H2 H3
CHO
Conversion
Modified prion protein PrPSc
Truncated prion protein PrP 27-30
CHO CHO
CHO CHO
Proteinase K
+
209aa
~142aa
N-liked carbohydrate chains
CHO
H1 Helical regions of PrPc
c
GPI (glycosylphosphatidylinositol)
b sheets in PrP
Repeats of 8 amino acids, P(Q/H)GGGWGQ
FIGURE 9.15 Isoforms of the human prion protein. The precursor protein is 254 amino acids long. Maturation involves
removal of the N-terminal signal sequence and the C-terminal 23 amino acids (two boxes marked S) N-linked glycosylation
at Asn181 and Asn197, and linkage of GPI near the new C terminus. After exposure to scrapie prions, the protein is converted
to PrPSc, which is partially resistant to proteinase K. This conversion involves loss of some helical regions (H's) in the cellular
form, and formation of new β sheets. Adapted from Weissmann (1996), Riek et al. (1996), and Prusiner (1998).
Ex Vivo and in Vitro Studies
displayed on the surface of the cell with a half-life of about 5
hours. It is then recycled into endosomal compartments and
Cell lines have been established that are persistently
eventually into lysosomes, where it is degraded.
infected with scrapie. These cells continuously produce
The function of PrPc is unknown. It is expressed by a
PrPSc, which allows biochemical studies to be performed
number of different cells, including neurons, hematopoietic
over a shorter time frame. The infected cells produce infec-
stem cells, and follicular dendritic cells. Many knockout mice
tious material that causes scrapie when inoculated into mice.
that lack the gene for PrP have been constructed and most
Of great interest has been the development of an in vitro
appear normal. The conservation of this protein indicates
system for the conversion of PrPc to PrPSc. In this system,
that it must perform some important function, but apparently
radioactive PrPc is mixed with unlabeled PrPSc, and the con-
its functions can be replaced by other proteins through the
version of the labeled PrPc to PrPSc is followed by its becom-
redundancy of many mammalian pathways. Recent studies
ing resistant to protease. These studies make clear that PrPc
have suggested that the protein is important for the renewal
can be converted to PrPSc by exposure to PrPSc in a process
of hematopoietic stem cells and for the development of neu-
that does not require the activities of intact cells. However,
rons, and that when an organism is stressed this develop-
so much infectivity is associated with the PrPSc added to the
mental function becomes critical.
reaction mixture that no increase in infectivity can be dem-
The brains of most humans or experimental animals
onstrated. Thus, these studies do not address the question of
exhibiting TSEs contain a conformational variant of PrPc
the nature of the infectious agent. These studies have also
called PrPSc (Sc for scrapie) or PrPres (res for resistant to pro-
been useful in the study of the species barrier, which can be
tease). PrPSc is found in aggregates that are largely resistant
quantitatively examined in such reactions.
to digestion with proteases. Treatment of PrPSc with pro-
teases and subsequent disaggregation of the proteolyzed
PrPSc give rise to a molecule that is truncated by about 80
Protein-Only Hypothesis
amino acids at its amino terminus (Fig. 9.15). In contrast,
It is clear that PrP is important in the development of
PrPc is completely destroyed by such protease treatment,
TSEs. There are two unresolved questions about PrP and the
and the normal PrP is also referred to as PrPsen (for sensitive
disease process, however. First, is the infectious agent that
to protease). Circular dichroism and infrared spectroscopy
indicate that PrPSc has a much higher content of β sheet than
leads to TSE PrPSc itself or is it another entity, such as a
does PrPc, 43 versus 3%, and a lower content of α helix,
virus? Second, does PrPSc, or some other modified form of
PrP, cause the symptoms of the disease, or is it simply a side
30 versus 42%, suggesting a profound conformational rear-
effect of the disease process?
rangement of the prion protein in the process of conversion
Preparations of the infectious agent purified from
from PrPc to PrPSc.
scrapie-infected mouse brain consist largely of PrPSc. There
is very little nucleic acid in infectious preparations of PrPSc.
Studies with Mice
In particular, there is no homogeneous DNA or RNA mole-
cule that might arise from a virus, for example. This has led
Transgenic mice have been useful in the study of TSEs.
to the hypothesis that PrPSc is itself the infectious agent. In
Mice have been made that lack the gene for the prion protein,
this model, "infectious" PrPSc induces PrPc to assume the
or that express wild-type or mutant prion proteins at levels
PrPSc conformation, and the accumulation of PrPSc in the
from less than normal to several times normal. Most mice
brain leads to the pathology associated with TSEs. Most of
that make no PrPc are normal, as described. However, such
the experimental data are compatible with such a model.
mice are resistant to scrapie infection. They do not become
PrPSc does induce PrPc to assume the PrPSc conformation, as
ill, and no infectious material is produced in the brain after
described earlier. Mutations in PrPc could make it easier for
inoculation of scrapie. In contrast, mice that overexpress
the protein to assume the PrPSc conformation, compatible
PrP are more sensitive to infection with scrapie. The incuba-
with the observation that some mutations result in inheritance
tion period is shorter, and the animals die more quickly after
of TSEs. The species barrier could result from lowered
inoculation with scrapie.
interaction affinities between proteins of different sequence.
Thus, the presence of PrP is essential for the development
However, it has not been possible to prove this hypothesis.
of TSE in mice. It has also been found that individual neu-
PrPSc preparations have a very low specific infectivity, with
rons must be able to produce PrP if they are to be sensitive
at least 105 molecules of PrPSc required for infection. Thus it
to scrapie-induced death. Neurografts from a donor mouse
remains possible that contaminants in the preparation might
that expresses PrP have been implanted into mice that lack
be required for infectivity. It has not been possible to dem-
the PrP gene. Upon inoculation of scrapie into the brain,
onstrate an increase in infectivity associated with the con-
the neurons in the graft develop a typical scrapie-induced
version of PrPc to PrPSc, as described before, which would
disease pathology. However, neurons outside the graft
provide solid evidence that PrPSc is infectious.
remain healthy.
In addition to the inability to prove the protein-only
Prusiner, was awarded the 1997 Nobel Prize for his "discov-
hypothesis, which could be due to the technical difficulties
ery of prions."
associated with this system, there are specific conceptual
difficulties with PrPSc as the infectious agent. One of the
Transport of Infectivity to the Brain
major criticisms of the protein-only hypothesis is the fact that
as many as 20 different strains of scrapie exist as assayed
Related to the conceptual problem of an infectious pro-
in mice. These strains of scrapie differ in properties such as
tein is how it might be transported to the brain after ingestion
the length of the incubation period following infection before
with food. This problem has been addressed in studies that
ascertain in which tissues PrPSc is present following inges-
disease becomes apparent, the areas of the brain affected, and
tion of PrPSc, and studies with transgenic mice that express
the symptoms of the disease, but these properties do not vary
within a strain. Such properties are expected for an infectious
PrP only in certain tissues. These various studies are com-
entity with a nucleic acid genome, but are difficult to recon-
patible with a model in which infectivity is transported via
cile with the properties of an infectious protein. If the protein-
axons following direct neuroinvasion of peripheral nerves.
only hypothesis is true, these differences in properties could
In the case of infection with only low doses of infectious
only result from differences in the conformation of PrPSc
material, amplification in follicular dendritic cells in lym-
in the different strains. How is it that a single, fairly small
phoid tissue may be required before neuroinvasion occurs.
Thus, in terms of the protein-only model, PrPSc might induce
protein can take up so many different conformations and that
the conversion of PrPc to PrPSc in cells in Peyer's patches,
each can induce the production of more protein having the
same conformation?
which then spreads via lymphatic tissue to peripheral nerves
Supporters of the protein-only hypothesis suggest that a
by sequential conversion of PrP.
limited number of conformational states of the prion protein
would be sufficient to explain the multiple strains of scrapie
For mation of the PrPSc Seed
that exist. They point to experimental data that show that at
If PrPSc can transmit the disease to a new susceptible host,
least two demonstrably different conformational states of the
prion proteins of two different mammals exist that "breed
how is it formed in the first place? Current models propose that
the conformational change resulting in PrPSc occurs rarely, but
true." Two different strains of transmissible mink encepha-
that once PrPSc is formed, it acts as a seed to induce the forma-
lopathy that produced different disease characteristics in mink
were passaged in hamsters. The PrPSc from the two strains,
tion of more PrPSc. Two models to explain the conversion of
PrPc to PrPSc by PrPSc have been proposed. In one, PrPSc (which
isolated from infected brain, are differently truncated at the
may be present in an aggregate) and PrPc form a complex, and
amino terminus on treatment with proteases in vitro. Thus the
conformations of the PrPSc in these two strains, both derived
the PrPSc induces the conformational change in PrPc. In the sec-
ond model, PrPc undergoes spontaneous transitions to different
from hamster PrP, must be different. Furthermore, this differ-
ence can be reproduced in an in vitro reaction in which PrPc
conformational states that are short lived and revert quickly
is mixed with the two different types of PrPSc. Each type of
to the native PrPc conformational state. These conformational
PrPSc induces PrPc to assume its own distinct conformation,
variants, however, can be locked into place by interaction
with PrPSc. In either case, the altered PrP joins the aggregated
as shown by the protease resistant fragment that is produced
from the PrPc on it conversion to PrPSc.
PrPSc to form a larger aggregate. Since the aggregated PrPSc is
In a second example of demonstrably different prion con-
insoluble, the reaction is essentially irreversible. Such a proc-
formations, human prions isolated from two different cases
ess could also explain the species barrier. The PrP proteins of
different animals differ slightly in sequence. PrPc that is identi-
of TSE, one FFI and the second CJD, were found to be dif-
cal in sequence to the PrPSc seed could interact with such a seed
ferently truncated after protease treatment. Passage of these
more readily than with a PrPSc seed that differs in sequence.
TSEs in transgenic mice that expressed a chimeric mouse
The protein-only hypothesis still requires a seed of PrPSc
human prion protein gave rise to prions in infected brain that
reproduced the differences in truncation. Thus, these confor-
to begin the reaction. One possibility is that it can form spon-
mational differences breed true when passed in mice.
taneously with a very low probability. Perhaps spontaneous
These studies demonstrate that PrPSc can exist in at least
changes in the conformation of PrPc to the PrPSc conforma-
two conformational forms, that the different conformational
tion might be fixed if this change occurred simultaneously
forms can produce different symptoms, and that the different
in a number of adjacent or interacting molecules. The effect
forms are capable of propagation by inducing PrPc to take up
of mutations in PrPc might be to increase the probability of
change to the PrPSc conformation, with the result that disease
their own particular conformation. Thus, the experimental
data are consistent with the protein-only hypothesis, although
occurs more frequently. Such a model is compatible with
it has not been proven conclusively and many still doubt its
data for human TSEs, where sporadic CJD occurs, albeit
validity. The hypothesis received a vote of confidence when
infrequently. However, sporadic disease has not been seen in
its most outspoken and passionate advocate, Dr. Stanley
shorter-lived animals. No sporadic BSE has been described,
can be killed by exposure to PrPSc. Thus simple neurotoxic-
and in countries where scrapie in sheep has been eradicated,
ity of PrPSc is not the cause of neuronal death. However, it
such as New Zealand and Australia, no recurrence of disease
is possible that conversion of PrPc to PrPSc at the surface of
has been observed.
the cell, which is known to occur, followed by accumulation
of PrPSc in lysosomes as the neuron attempts to recycle it,
Does PrPSc Cause the Disease?
could be toxic. In this model, it is the resistance of PrPSc to
If PrPSc is responsible for the pathology of TSE disease,
proteases in the lysosome that results in toxicity.
and not simply a by-product of disease, the mechanism by
Recent findings have suggested another possibility. PrP
which it causes disease is uncertain. An early model sug-
can be expressed as a membrane-spanning protein as well as
gested that PrPSc itself is neurotoxic. However, it has been
a protein anchored by a glycolipid anchor (Fig. 9.16). One
shown that a neuron must be able to express PrPc before it
membrane-spanning form, called CtmPrP, has its C terminus
Conformations of the human prion protein translated in vitro
A.
N N
S
181 197 231
STE TMI
N S
S C
E1
E2
C N
N
C
Lumen
Microsomal
membrane
Cytosol
N
C
CtmPrP
secPrP
NtmPrP
B.
Maturation of secPrP in cells
N S
N
N
Transport through
Maturation
post-ER compartments
C
S
Lumen
Lumen
ER
ER
PM
Cytosol
Cytosol
Cytosol
secPrP
PrPc
PrPc
STE--stop transfer effector
N-linked carbohydrate chains
TMI--transmembrane domain
GPI-glycosylphosphatidylinositol
E1 (epitope for MAb 3F4)
Repeats of 8 amino acids
E2 (epitope for MAb 13A5)
FIGURE 9.16 Postulated topology of PrP proteins in membranes. (A) Topology of PrP proteins in membranes after
translation in a cell-free system supplemented with microsomes. The topology was determined by a combination of protease
digestion from the cytosolic compartment and identification of the domains protected within the lumen using the two
MAbs, 3F4 and 13A5. Mutations have been shown to affect the ratio of the three forms shown, and greater concentrations
of CtmPrP are associated with neurodegenerative disease in mice. (B) Model for maturation and association with membranes
of SecPrP in cells. ER, endoplasmic reticulum; PM, plasma membrane. Adapted from Hegde et al. (1998).
Search WWH :