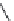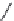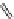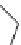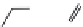Biomedical Engineering Reference
In-Depth Information
NH
NH
NH
NH
O
O
O
O
O
O
NH
O
O
O
O
CF
3
3.14
K
i
= 122 nM
3.16
K
i
= 20 nM
3.17
K
i
= 1.4 nM
3.18
K
i
= 0.26 nM
NH
NH
NH
O
O
NH
O
O
O
3.17
K
i
= 1.4 nM
3.19
K
i
= 0.17 nM
3.20
K
i
= 0.048 nM
FIGURE 3.11
Pharmacophore-guided optimization of compound
3.14.
The replacement of the CF
3
group in
3.14
by an ethyl group to give
3.16
increases the afi nity
from 122 to 20 nM. Replacement of the ester ethyl group in
3.16
by a propyl group to give
3.17
further increases the afi nity to 1.4 nM and conversion of the ester group in
3.17
to an amide group
gives compound
3.18
with an afi nity of 0.26 nM.
By further exploring lipophilic substituents in the 3
-position, compound
3.19
was identii ed to
be a high afi nity compound (
K
i
= 0.17 nM). Finally, converting the ester group in
3.19
to an amide
group gave the highest afi nity compound
3.20
in this series of compounds with
K
i
= 0.048 nM. In
comparison with compound
3.14
, compound
3.20
has a higher afi nity by a factor of 2500. This
increased afi nity demonstrates the power of a well-developed pharmacophore model for the opti-
mization of a compound with respect to afi nity.
′
3.7 3D-QSAR ANALYSIS—THE GRID/GOLPE APPROACH
QSAR methods in medicinal chemistry use statistical methods to correlate the variation in molecu-
lar properties with the variation in biological activities. The purpose of establishing a QSAR model
is to be able to predict activities of new compounds quantitatively. Traditional QSAR methods in
general do not explicitly take 3D structures into account, but use substituent parameters to describe
the variations in molecular structures/properties. In contrast, 3D-QSAR methods explicitly use
3D molecular structures and use molecular interaction i elds to describe the variation of the properties
of the molecules.
Several alternative approaches to 3D-QSAR have been developed. In this chapter, we illustrate
the 3D-QSAR methodology by the GRID/GOLPE approach.
3.7.1 GRID M
OLECULAR
I
NTERACTION
F
IELDS
Molecular interaction i elds describe interaction energies between a molecule and a chemical probe
positioned in different locations around the molecule. GRID is a widely used program for the cal-
culation of such i elds. The chemical probe may be a methyl group, a water molecule, or any of
the more than 60 probes provided by GRID. Interaction energies between the molecule and the
probe are calculated by inserting the molecule in a box (Figure 3.12). The probe is then moved
through a regular 3D array of grid points at positions around the molecule as shown in the i gure.
The spacing between the grid points is user-dei ned but normally 0.25-1Å. At each grid point the