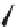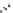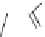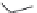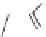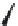Biomedical Engineering Reference
In-Depth Information
F
OH
OH
OH
OH
F
OH
H
2
N
H
2
N
H
2
N
O
O
O
O
O
H
2
N
H
2
N
F
(
15.4
)
Vigabatrin (
15.3
)
(
15.5
)
(
15.6
)
(
15.7
)
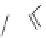
FIGURE 15.2
Structures of some GABA-AT inhibitors, compound
15.6
being inactive.
(Figure 15.2), a suicide inhibitor for the enzyme GABA-aminotransferase (GABA-AT) responsible
for GABA degradation, is used clinically as an anticonvulsant. Elevation in extracellular GABA
levels by the inhibition of the reuptake of GABA is effected by Tiagabine (
15.20
) (refer to
Figure 15.4) marketed for the treatment of epilepsy and in preclinical studies for treatment of
anxiety and insomnia. The G-protein-coupled GABA
B
receptor is the target for the antispastic drug
baclofen (
15.41
) (refer to Figure 15.9).
Drugs targeting the Glu neurotransmitter system have been slower to emerge. Memantine (see
Section 15.7.3) is used with some success for treatment of Alzheimer's disease and a few com-
pounds with mixed mechanisms of action, including reduction of Glu release (through blockade of
Na channels) are used for the treatment of migraine, epilepsy, and amyotrophic lateral sclerosis.
All of the Glu and GABA receptors and transporters are heterogeneous and may individually be
involved in specii c CNS disorders and disease conditions. These receptor/transporter subtypes are
unevenly distributed in the CNS, which opens up the prospect of developing ligands selective for
receptor/transporter subtypes with predominant location in different brain regions of therapeutic
relevance.
15.3 GABA BIOSYNTHESIS AND METABOLISM
The GABA concentration is regulated by two pyridoxal 5
-phosphate (PLP) dependent enzymes,
l-glutamic acid decarboxylase (GAD), which catalyzes the decarboxylation of Glu to GABA prior
to release into the synaptic cleft, and GABA-AT, which degrades reuptaken GABA to SSA (Figure
15.1). This transamination step takes place within presynaptic GABA terminals as well as in sur-
rounding glia cells.
′
15.3.1 I
NHIBITORS
OF
GABA M
ETABOLISM
A number of mechanism-based inactivators of GABA-AT has been developed and has been shown
to elevate the extracellular levels of GABA. These compounds are typically analogs of GABA, con-
taining appropriate functional groups at C4 of the GABA backbone (e.g.,
15.3
,
15.4
, and
15.5
). The
functional group is converted by GABA-AT into electrophiles, which react with nucleophilic groups
at or near the active site of the enzyme and thereby inactivate the enzyme irreversibly (Figure 15.3).
The most effective of these,
γ
-vinyl-GABA (Vigabatrin,
15.3
), is clinically used as an anticonvulsant
for the treatment of epilepsy.
The mechanism for inactivation of GABA-AT by Vigabatrin (
15.3
) is outlined in Figure 15.3. As
shown, PLP is forming a Schiff base (
15.8
) with the terminal amino group of a lysine residue in the
active site of GABA-AT. Transamination with
15.3
generates a new imine
15.9
, which undergoes a rate-
determining enzyme-catalyzed deprotonation to give the imine
15.11
after reprotonation. In analogy
with transamination reaction on GABA,
15.11
could be hydrolyzed to give the SAA analog
15.13
and
pyridoxamine-5-phosphate
15.12
. However,
15.11
is a Michael acceptor electrophile, which undergoes
conjugate addition by an active-site nucleophile X and the inactivated enzyme
15.14
is produced.
To optimize the effect of Vigabatrin a number of conformationally restricted GABA analogs has
been developed. In contrast to the Vigabatrin analog
15.6
, which does not inactivate GABA-AT, the