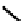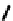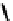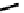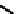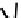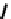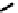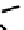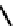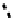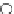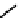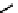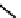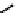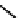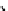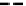Chemistry Reference
In-Depth Information
N
R
2
R
+
R
R
2
N
Tc
P
S
P
Cl
R
1
R
1
MeN
S
N
Re
P
R
2
N
P
Cl
R
2
R
1
R
R
R
1
=CH
2
CH
2
OEt
R
2
=CH
2
CH
2
CH
2
OMe
40
R=alkyl or aryl
39
fIgure 6.13
nitride complexes with aminophosphine ligands.
R
Ph
R
MN
N
MNN
MNN
H
H
Diazenide 1
-
Hydrazide 2
-
Isodiazene 0
41
42
NHR
M H
2
M H
HN
M
R
HN
R
NH
H
1
-hydrazide 1
-
H
2
-hydrazide 1
-
Hydrazine 0
43
44
fIgure 6.14
Structures and bonding in diazenide, isodiazene, hydrazide, and hydrazine complexes.
For maximum flexibility in deploying the nitride core, it would be desirable to synthesise complexes with a mixed ligand
system analogous to the 3 + 2 systems described above for oxo complexes. Unfortunately, the same approach for the nitrides
gives mixtures of compounds. However, it has been shown that the stereochemistry and stoichiometry of the Tc or Re nitride
core co-ligands can be neatly controlled by the use of tridentate PnP donor ligands where the π-acceptor P donors prefer to
be
trans
. This forces the halide ligands to be
cis
-oriented, and they can readily be substituted by a range of bidentate
monoanionic or dianionic ligands [136]. The structure of the Re dichloride (
39
) is shown together with that of a cationic
dithiocarbamate derivative (Tc-n-DBDoC,
40
), which shows promise as a cardiac imaging agent
in vivo
in both animals and
humans [137]. The selection of other functionalisable bidentate ligands enables these complexes to be conjugated to a range
of biomolecules [138-142].
6.2.4
diazenide, Isodiazene, and hydrazido complexes
The bonding and formal charges adopted by this group of ligands are much less straightforward than for the oxo and nitride
ligands considered hitherto, and a further complexity is that they can be interconverted via protic equilibria (Figure 6.14).
Structure
41
shows the most common bonding mode for the 'singly bent' diazenide ligand where it bears a monoanionic
charge. Diazenide complexes can be prepared by a number of routes, but those using mono-substituted hydrazines or diazo-
nium salts have been the most prevalent. The R group can be alkyl, aryl, or aroyl. Protonation at the diazenide nitrogen
remote from the metal produces the mnnHR ligand
42,
which generally has a linear m-n-n system. Two canonical forms
(hydrazide(2-) and isodiazene) can be drawn differing in the distribution of electrons in the m-n and n-n bonds. There is
not a simple VB representation that covers the multicentre σ and π bonding system in the isodiazene form, thus dotted bonds
have been used. X-ray crystal structures of complexes with nnR
2
ligands show an n-n distance more appropriate for a
double rather than single bond; therefore, the formal nomenclature used for these ligands is isodiazene. However, it should
be noted that this is solely for the purposes of providing a name and should not be interpreted that these ligands confer a
neutral charge. In general, it is difficult to assign a true metal oxidation state to complexes of ligands that can undergo facile
electron redistribution. If there is a donor group on the diazenide substituent (as in HynIC, shown in Figure 6.15), then the
m-n-n system is no longer linear and rehybridisation of the nitrogen adjacent to the metal from sp to sp
2
permits protonation
to occur using the lone pair now available. Addition of a further proton to the nitrogen of isodiazene remote from the metal
produces the hydrazido(1
−
) ligand
43,
which can bind end-on or side-on to the metal. A third protic addition gives the neutral
hydrazine ligand
44
. note that the positive charges that would accompany protonation have been omitted for clarity.