Biology Reference
In-Depth Information
provide an important clinical basis for a link between
DNA repair and neuronal function. Given the number
of non-neuronal targets that could be affected by such
mutations, however, the compromised repair could
affect systems that, in turn, indirectly affect neuronal
function. Second, assuming that augmenting DNA
repair will attenuate neurotoxicity induced by cancer
therapies, studies will be needed to show that such
manipulations do not interfere with the anticancer
actions of the therapy.
Recent studies have attempted to establish a causal
relationship between altering DNA repair and neuro-
toxicity caused by oxidative DNA damage or by
therapy-induced neurotoxicity. These studies examine
neurotoxicity in transgenic animals with deficiencies
in DNA repair proteins or after manipulations of
expression of repair proteins. To date, most of these
studies focus on the BER pathway since it is the major
repair pathway for oxidative DNA damage in the
nervous system.
13
Twomajorstepsinthispathway
are the removal of the damaged bases by glycosylases
such as oxoguanine 8-hydroxyguanine glycosylase
(OGG1), which excises 8-oxo-guanisines and leaves
an AP site.
183
The phosphodiester backbone 5' at this
site is then hydrolyzed by APE1, generating a normal
3'-hydroxyl group and a deoxyribose-5-phosphate.
184
We and others have examined the effect of altering
the BER on neurotoxicity induced by oxidative stress.
The relevance of these studies to cancer-induced neuro-
toxicity is based on the premise that oxidative DNA
damage is a major mechanism for neuronal damage
induced by ionizing radiation and anticancer drugs.
Using transgenic mice with a deficiency in OGG1, Liu
and co-workers showed that exposing cortical neurons
from these mice to 40
response to oxidative DNA damage. Overexpression
of APE1 also is neuroprotective after ischemic injury
to the central nervous system. In rats with a global
ischemic injury, the expression of APE1 in the hippo-
campus is reduced, and this reduction correlates with
decreased DNA repair and increased cell death in
CA1 neurons.
187
Administration of pituitary adenylate
cyclase-activating polypeptide (PACAP) intracerebrally
induces APE1 expression and activity in the hippo-
campus of rats and suppresses DNA damage in hippo-
campal neurons produced by global ischemia.
187
This
peptide also reduces the phosphorylation of the histone
H2AX in hippocampal neurons, suggesting that it
reduces DNA double-strand breaks. Injecting a lentivi-
ral construct containing APE1shRNA into the CA1
region of the rat hippocampus significantly reduced
APE1 expression and blocked the neuroprotective effect
of PACAP.
187
Overexpression of an APE1 mutant that
lacks DNA repair capabilities did not prevent
ischemia-induced DNA damage. These results provide
(A)
(B)
MH
2
O
2
for 24 hours resulted in
a significant increase in cell death 6 hours after the insult
compared to neurons harvested from wild-type mice.
185
Using focal ischemia by occluding the left middle cere-
bral artery, they observed an increase in the size of the
infarct and an increase in oxidative DNA damage in
the brains of mice deficient
m
in OGG1compared to
controls.
Alterations in the expression and activity of the
endonuclease APE1 also affect the degree of oxidative
DNA damage in neuronal tissues. In isolated rat
cortical neurons grown in culture, exposure to 20
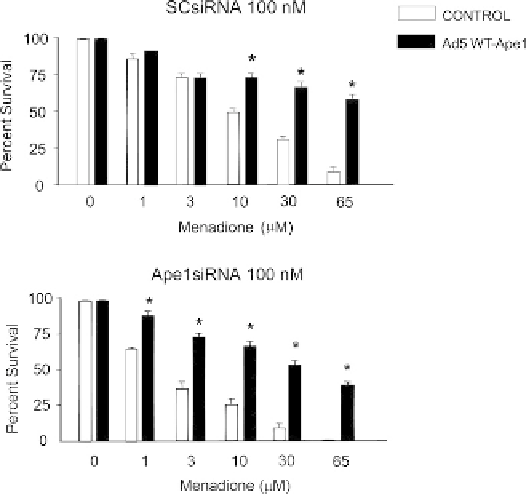
FIGURE 13.2
Menadione-induced cell death in sensory neuronal
cultures is increased by a reduction in APE1 expression and attenu-
ated by APE1 overexpression. Sensory neuronal cultures grown for 11
days were exposed to various concentrations of menadione for 1 hour.
The cultures were pretreated with (A) scramble siRNA (SCsiRNA) or
(B) APE1siRNA on days 4
M
glutamate for 10 min causes oxidative DNA damage
and increases the expression and activity of APE1
when measured 6 hours after treatment.
186
Although
this damage and the increase in APE1 expression return
to control levels within 24 hours, when the expression
of APE1 is reduced using small interfering RNA
(siRNA), the neuronal DNA damage is still observed
at 24 hours after the glutamate treatment. These results
suggest that the BER pathway can be mobilized in
m
6 in culture and infected for 24 hours on
day 8 with an adenoviral construct containing the CMV promoter and
EGFP (vector control; open columns) or a construct containing the
CMV promoter, human APE1, IRES, and EGFP (Ad5 WT-APE1;
shaded columns). Each column represents the mean
e
SEM of the
percent of cell viability of three independent harvests. Viability is
measured by trypan blue exclusion 24 hours after removing the
menadione. An asterisk indicates a statistically significant difference
in vector-treated versus Ad5-WT-APE1-treated cultures using analysis
of variance and the Tukey post-hoc test.