Biology Reference
In-Depth Information
bifunctional alkylating drugs and platinum compounds
(reviewed in
97,243
). However, predictions of drug sensi-
tivity are complicated by several factors. First, there are
differences in the nature of the distortion introduced
into DNA by ICLs.
243
For example, mitomycin C creates
little distortion of the DNA helix compared to nitrogen
mustards or cisplatin, which may affect lesion recogni-
tion and repair. Second, these drugs do not produce
only one defined type of damage but a spectrum of
lesions, all of which may impact drug sensitivity. It is
often overlooked that for mitomycin C the fraction of
ICLs produced appears by far to exceed the relative
amount of intrastrand adducts.
244
In contrast, the
damage spectrum after cisplatin treatment is dominated
by intrastrand adducts while ICL comprise only less
than 10% of the total.
97,245
Thus, the requirement for
certain HRR components may be different for mito-
mycin C versus cisplatin.
163
It also remains unknown
whether intrastrand adducts or ICL represent the critical
lesion of cisplatin mediated cytotoxicity.
246,247
The
pronounced cisplatin sensitivity of HRR-deficient cells
(such as XRCC2/3 or BRCA1) would argue for an
impaired ability to cope with ICL, even though levels
are relatively low, or to restart forks following collision
with persistent intrastrand adducts that have escaped
NER.
246,248
Third, removal of ICL requires a complex
interaction between multiple repair pathways and
checkpoint responses,
243
so that the activity of a single
pathway such as HRR may not necessarily predict the
entire degree of cell kill. Lastly, it needs to be borne in
mind that many agents are subject to redox biotransfor-
mation, which may influence their toxicity in cells defi-
cient for components of the FA or HRR pathways.
164,166
The concept that HRR is required to cope with lesions
that block advancing replication forks extends to several
other types of agents (
Tabl e 7 .2
) as well as PARP1 inhibi-
tors which are dealt with elsewhere (see Chapter 4, and
below). For example, topoisomerase I functions to over-
come the winding problem ahead of replication forks
by inducing a transient SSB. Upon topoisomerase I inhibi-
tion, forks will run into the persistent SSB, collapse, and
trigger HRR.
249
HRR-deficient tumors are thus expected
to be hypersensitive to this class of agents. Topoisomerase
II poisons, such as etoposide or doxorubicin, may also
create fork-blocking DSB as well as post-replication
breaks, both of which require proficient HRR.
250
Helleday and colleagues discovered that 6-
thioguanine, a thiopurine, killed HRR defective breast
cancer cells as effective as a PARP inhibitor.
251
The precise
mechanisms for this phenomenon remain to be estab-
lished but likely involve HRR for the repair of 6-
thioguanine-induced secondary DSB as well as other
recombinogenic lesions. These findings have inspired
a new clinical trial to treat BRCA defective cancers with
6-thioguanine prodrug.
242
O
6
-methylguanine is an adduct
(A)
(C)
(B)
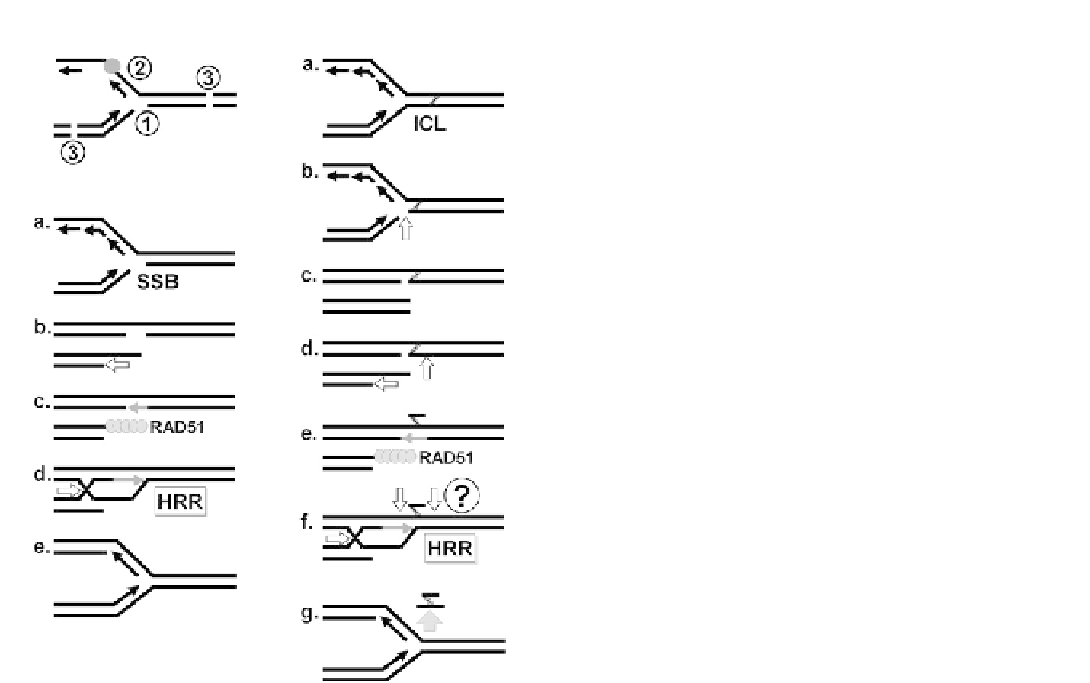
FIGURE 7.8
Models of damage processing and repair at replica-
tion forks. (A) Types of damage at replication forks (based on Helle-
day
et al.
91
): (1) Postulated one-ended DSB generated upon collision of
replication fork with, for example, a single-strand break (SSB) or
interstrand crosslink (ICL). (2) Daughter strand gap is generated when
leading or lagging strand synthesis is blocked by a single-stranded
lesion. (3) Two-ended DSB ahead of a replication fork or in a sister
chromatid. (B) Upon encountering an unrepaired SSB, for example
due to PARP inhibition, the replication fork collapses (replication run-
off) and a one-ended DSB is created (based on Helleday
et al.
91
).
Following resection of the end (indicated by arrow), invasion of the 3
0
end into the sister chromatid is accompanied by D-loop formation.
Cleavage of the Holliday junction restores the replication fork. (C) The
replication fork stalls at an ICL and a one-ended DSB is created by the
MUS81 endonuclease (arrow) (based on Ciccia and Elledge,
1
Helleday
et al.
97
). This is followed by ICL unhooking and processing of the
double-stranded end (arrows). The single-stranded gap is closed by
TLS. It is not established how exactly HRR and NER cooperate to
complete ICL repair (illustrated by a question mark). Though we
show here only the presence of a single fork, some recent models, for
example by Ciccia and Elledge,
1
invoke the arrival of a second fork to
facilitate HRR. However, the likelihood by which two forks stall at
a single ICL in the context of replication checkpoint activation is
unknown. Either way, it is thought that strand invasion and RAD51-
mediated synthesis-dependent strand annealing are followed by
removal of the crosslink by NER as ERCC1 has been recently shown to
function downstream of RAD51.
Role of HRR for the Repair
of Chemotherapy-Induced DNA Lesions
Typically, HRR-deficient cells are most sensitive to
agents that generate replication fork-blocking ICL, i.e.,