Biology Reference
In-Depth Information
they are highly unlikely to eliminate conventional
DNA damaging therapies in the foreseeable future.
Moreover, the detection of HRR and other DNA repair
defects in many cancers has reinvigorated efforts to
“target” these repair pathways with treatment
approaches based on conventional DNA damaging
agents. Below, we will review major treatment strategies
that can be employed to either exploit pre-existing HRR
defects in cancers or pharmacologically disrupt intact
HRR, with the goal of increasing the effectiveness of
conventional chemotherapeutics as well as radiation in
subsets of cancer patients.
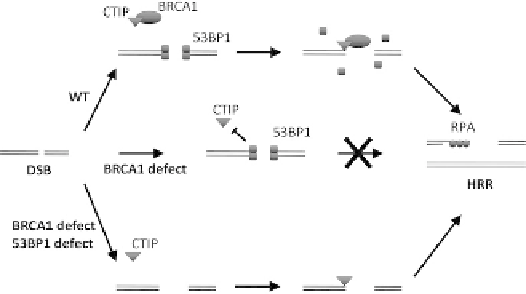
FIGURE 7.6
Model of BRCA1
0
s and 53BP1
0
s involvement in HRR
control. This model for the functional interplay of BRCA1 and 53BP1
in controlling end resection and HRR initiation is derived from the
work of Nussenzweig and colleagues.
89
In normal cells (WT, wild-
type), BRCA1 functions to remove 53BP1 from double-stranded ends,
thereby allowing ATM/CtIP-dependent strand resection, RPA
loading, and subsequent HRR initiation. In the absence of BRCA1,
53BP1 protein is not displaced, HRR cannot be initiated, and the break
persists which can lead to chromosomal aberrations. Additional loss
of 53BP1 rescues the cell's inability to initiate HRR in the absence of
BRCA1 by allowing CtIP access to the ends and strand resection.
Targeting HRR Defects with DNA
Damaging Anticancer Agents
A large number of chemotherapeutic agents
commonly used in cancer therapy cause DNA lesions
that are known or expected to require HRR for cell
survival (
Figure 7.7
).
92,241,242
Typically, HRR is needed
for replication fork repair and restart after the fork has
encountered blocking lesions such ICL, SSB, or topoiso-
merase II-associated DSB. It has become popular to illus-
trate this concept as shown in
Figure 7.8
.
91,97
However, it
is important to point out that it is still poorly understood
how exactly HRR occurs at stalled or collapsed replica-
tion forks in mammalian cells (see above). HRR is also
required for homology-mediated repair of post-replica-
tion DSB in cells in late S/G2 phase of the cell cycle.
Thus, tumors with HRR defects are likely sensitive to
a large variety of chemotherapeutic agents.
Table 7.2
provides an overview of some of the common chemo-
therapeutic agents, the type of DNA damage caused
by these, and the role of HRR pathways in the repair
of damage. Some less commonly used agents that have
attracted renewed interest due to an increasing appreci-
ation of HRR defects in cancer are also included.
about 15% of all breast cancers.
88
In another study, 26%
of breast tumors were found to have reduced or absent
53BP1 expression on immunohistochemistry, similar to
24% of lung cancers, though whether these observations
correlate with an underlying HRR defect
is not
known.
189
Conclusions
From the observations summarized above it can be
concluded that defects in HRR regulatory and mediator
proteins may be much more common in human cancers
than previously appreciated, but even though they are
associated with genomic instability they do not neces-
sarily confer sensitivity to DNA damaging agents due
to compensatory increases in HRR.
129
Therefore, testing
for the status of a single HHR gene, such as BRCA1, may
not provide an appropriate measure of HRR activity in
a given tumor without also assessing for the presence
of compensatory mechanisms such as RAD51 overex-
pression, p53 mutation, 53BP1 loss, or other yet unchar-
acterized alterations.
STRATEGIES TO TARGET
HRR IN CANCER
Genotoxic chemotherapeutics and radiation therapy
constitute the mainstays of treatment for most human
cancers. While molecular targeted drugs, such as imati-
nib in chronic myeloid leukemia or EGFR tyrosine
kinase inhibitors in subtypes of lung adenocarcinoma,
have introduced a paradigm shift in cancer therapy,
FIGURE 7.7
Illustration of cell-cycle specific HRR in response to
DNA damaging agents.