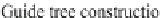Biology Reference
In-Depth Information
Fig. 1 Diagram of the program workflow
refinement can be performed as a post-processing step of stage (5) to
further improve alignment accuracy. Figure
1
shows the diagram of
the program workflow.
Given two sequences
X
and
Y
, define
X
i
to denote the
i
th residue in
X
and
Y
j
to denote the
j
th residue in
Y
. Assuming
A
to be the space
of all possible global alignments of
X
and
Y
and
a
*
3.2 Pair Hidden
Markov Model
A
be the
“
true
” alignment of the two sequences, the posterior probability
that
X
i
is aligned to
Y
j
(denoted as
X
i
~
Y
j
)in
a
*
is defined as
2
X
a
j
P
ð
X
i
Y
j
2
X
;
Y
Þ¼
P
ð
a
j
X
;
Y
Þδf
X
i
Y
j
2
a
g;
(1)
a
2
A
{
cond
}
returns 1 if the condition
cond
is true and 0, otherwise.
P
(
a
|
X
,
Y
)
represents the probability that
a
is the
true
alignment
a
*
.Thus,
P
(
X
i
~
Y
j
2
i
|
X
|and1
j
|
Y
|. The indicator function
where 1
δ
a
*
|
X,Y
), i.e.,
P
(
X
i
~
Y
j
) for short, can be considered
as the probability that
X
i
is aligned to
Y
j
in the true alignment
a
*
.
The posterior probability matrix
P
XY
of
X
and
Y
is a two-
dimensional
table of
size |
X
|
|
Y
|, consisting of all values
P
(
X
i
~
Y
j
)for1
|
Y
|.
Figure
2
shows the used pair-HMM model to specify the
probability distribution over all alignments
A
of a sequence pair.
This pair-HMM model has three states:
M
,
I
, and
D
. At state
M
,
one residue is emitted for each of the sequences
X
and
Y
, meaning
that the two residues are aligned together. At state
I
, it only emits
one residue for sequence
X
, meaning that this residue from
X
is
aligned to a gap. Similarly, state
D
only emits one residue for
sequence
Y
, meaning that this residue from
Y
is aligned to a gap.
To compute the posterior probabilities, we used both the forward
and backward algorithms as described in [
3
].
i
|
X
|and1
j