Biology Reference
In-Depth Information
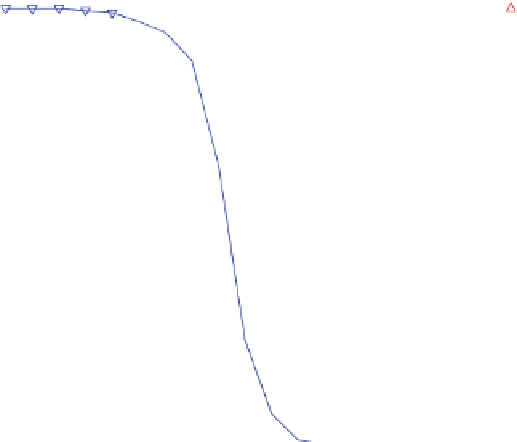
Fig. 4 Relative grammar-based distance classification thresholds at the genus level. Given a known set of 16S
Ribosomal RNA sequences in which multiple sequences belong to the same genera, a binary classification
is performed for varying grammar-based complexity thresholds. The classification procedure is to compare
the pairwise distance between two sequences against a threshold. If the distance is less than the
threshold, the two sequences are classified as belonging to the same genus. If the distance is greater than
the threshold, the two sequences are classified as belonging to different genera
actually calculated, as more similar sequences are less likely to
require the entire matrix be created.
Option -v <value>
: Depicted in Fig.
5
, specify the alignment
overhang percentage for similar sequences. Normally in pairwise
alignment every residue of one sequence is compared to all other
residues of the second sequence. Especially for nearly identical
sequences, this is unnecessary. This value represents the percent of
residues shifted past both ends of the longer sequence. Smaller
values will result in quicker alignments but at a risk of increased
mismatches. If this option is not specified, the default value is 0.10.
Suggestion:
This value can be lowered to decrease computation
time, but attention must be paid to the
-T
option. Increasing the
-T option will result in more dissimilar sequences being grouped
together, and then be pairwise aligned using this parameter. You
should only lower this value if you also lower the -T value as well.
Option -V <value>
: Similar to the previous option and depicted
in Fig.
6
, specify the alignment overhang percentage for dissimilar
sequences. The alignment of more dissimilar sequences will cause
the algorithm to deviate away from the diagonal of the matrix by a