Biology Reference
In-Depth Information
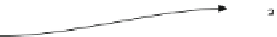
Fig. 1 Algorithm overview. Given the sequences to be aligned, {s
1
,
...
, s
6
}, a distance matrix is calculated
based on relative grammar complexity. The sequences are subsequently placed into similarity groups based
on their relative distance compared to a user-specified similarity threshold (option -T). Sequences within each
group are progressively aligned to form a group consensus sequence, cs
i
. Finally, the consensus sequences
are progressively aligned to result in the final alignment
phylogeny which in turn is required for determining the order in
which the sequences are aligned. Some alignment algorithms over-
come this by introducing a number of iterations to continuously
refine both the distance estimations and the alignment. As will be
described,
GRAMALIGN
generates a distance matrix based on a mea-
sure of relative grammar complexity naturally inherent in biological
sequences which does not require knowledge of the underlying
phylogeny. This section is dedicated to describing this computa-
tionally efficient way of creating a valid alignment order prior to
performing the progressive alignment.
Measures of distances between sequences are central to the devel-
opment of molecular phylogeny. The measures of distance can be
divided into two main classes, distances whose computation
requires that the sequences be aligned, and methods that can
compute distances without the need for alignment. Some of the
measures that require sequence alignment [
7
-
11
] use a nucleotide
or amino acid substitution model to develop a distance matrix
which is then used to build a phylogenetic tree. Others use parsi-
mony and maximum likelihood to evaluate the fitness of different
topologies for the phylogenetic trees [
12
-
19
]. All these approaches
assume the existence of homologous versions of a particular gene
in each of the organisms which need to be aligned in order for
the distances to be computed. The requirement of MSAs imposes a
substantial computational cost which can become prohibitive when
the number of sequences is large. Alignment free methods were first
introduced by Sankoff et al. [
20
] to deal with situations where the
comparisons were not with homologous genes but rather with whole
genomes. Since then a number of alignment free methods that
2.1 Phylogeny
Estimation