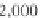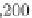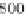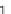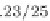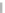Biology Reference
In-Depth Information
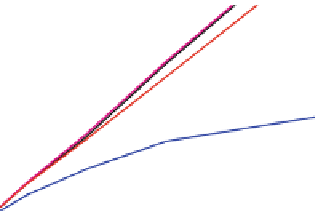
Fig. 2 The length of the alignment is expected to grow linearly with the evolutionary divergence contained
within the sequences. One thousand sequences were simulated under a random tree with the maximum root-
to-tip distance of 0.1 substitutions per site [
17
]. Subsets of 10, 25, 50, 100, 250, and 500 sequences as well
as the full datasets were re-aligned with ClustalW [
7
] and
PRANK
, and the length of the resulting alignments is
plotted as a function of the length of the tree relating the included sequences. As the insertion-deletion
process used for simulation is time-dependent and defined relative to the substitution rate, the correlation
between the two values for the true alignment (black line) is perfect. The two variants of the phylogeny-aware
function (
PRANK
and
PRANK
+F
) produce alignments with lengths close to the true length whereas a method based
on the classical progressive alignment algorithm (ClustalW) over-aligns the sequences and the length of the
alignment is seriously underestimated. Solid and dashed lines indicate alignments based on the true and
estimated guide trees, respectively. The rectangle in the left plot indicates the area shown on the right
considering the phylogeny of the sequences included. In practice,
not only are insertions at the same position problematic but the
correct alignment of sequences with insertions and deletions at
near-by positions requires the identification of distinct evolutionary
events and their subsequent correct handling.
Progressive alignment algorithms exploit the sequence phylog-
eny and align the sequences pairwise in the reverse order, starting
from the most closely-related ones and, at each step clustering the
aligned subsets, progressing towards the root of the tree. A major
reason for the use of progressive algorithms is the prohibitive
computational complexity of the exact multiple sequence align-
ment algorithm: with progressive algorithms,
the complexity
is reduced from
O
(
l
n
)to
of aligning
n
sequences of length
l
1)
l
2
). The additional beauty of the approach is that the
algorithm starts with alignments that are expected to be easiest and
thus minimizes the chances of early alignment errors in its greedy
processing of sequences. The classical algorithm does not use the
phylogeny for anything else, however, and the placement of gaps—
that is, the inference of which characters have been inserted or
deleted in the evolutionary past—in the resulting alignments is
often phylogenetically implausible [
5
].
O
((
n