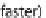Biology Reference
In-Depth Information
Table 1
Comparison of different options using the 16S.B.ALL dataset [
49
]
Actual time
{
Command
Accuracy CPU time
mafft
--addfragments frags existingmsa
0. 9969
6.67 days 18.3 h
mafft --6merpair --addfragments frags existingmsa
0. 9949
3.77 h
36.2 min
39.7 days
{
4.21 days
{
mafft --localpair --add
frags existingmsa
0. 9707
mafft --6merpair --add
frags existingmsa
0. 9604
1.32 h
1.44 h
profile alignment
0. 2779
14.8 h
1.53 h
The estimated alignments were compared with the CRW alignment to measure theaccuracy (the number of correctly
aligned letters/the number of aligned letters inthe CRW alignment). Calculations were performed by MAFFT version
6.954, on aLinux PC with 2.67 GHz Intel Xeon E7-8837/256 GB RAM (for the case marked with
{
), oron a Linux PC
with 3.47 GHz Intel Xeon X5690/48 GB RAM (for the othercases)
{
Wall-clocktime with ten cores. Command-line
argument for parallel processing is
--thread 10
[
42
]
sequence of the known species. However, in metagenomic analysis
when new sequences are from multiple (and some novel) species,
the phylogenetic position of the new sequences should be consid-
ered, like PaPaRa [
46
], PAGAN [
48
] and this option of MAFFT.
The accuracy of resulting MSAs was estimated by comparing
them with the original CRW alignment (Table
1
). CPU time and
wall-clock time for each method are also listed in the table. Since
the sequences in this dataset are highly conserved, the difference in
accuracy between the default (
--addfragments
) and the faster
option (
--6merpair --addfragments
) is small.
We also compared the performances of some subtypes of the
--
add
option using the same dataset.
These options have no advantage for this problem, according to the
third and fourth lines in Table
1
. This is probably because the
relationship among new fragments does not make sense,
since most of them do not overlap with each other. In such cases,
--addfragments
, which does not consider this relationship, is
more suitable than
--add
, which considers this relationship.
This observation suggests that the trade-off between accuracy and
speed does not always hold. Rather, a method designed for the
appropriate purpose should be applied. The application of a com-
putationally expensive method based on L-INS-1 (
--localpair
--add
) has no advantage, because the extra computational time is
spent on the comparison of non-overlapping fragmentary
sequences, which have no reasonable solutions.