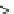Biology Reference
In-Depth Information
Moreover, biological knowledge is sometimes incorporated into
MSAs in databases. Such information can be retained in a large
alignment if the original alignment is kept. Based on such considera-
tions, around 2010, we implemented an option,
--add
, to add
unaligned sequences to an existing MSA. The implementation of
the
--add
option was almost trivial; no change was necessary from
the conventional progressive method, except that the alignment
calculation is skipped at the nodes whose children are all in the
existing alignment.
Several tools [
46
-
48
] for aligning short reads to existing align-
ment were developed between 2011 and 2012. Indeed such analy-
sis is recently becoming important, along with the popularization
of second-generation sequencers. For this purpose, a limitation of
the
--add
option of MAFFT was pointed out in [
48
]. Thus we
implemented a new option,
--addfragments
, which does not
consider the relationship among the sequences to be added, for
this purpose. Details of the
--add
and
--addfragments
options
are described in [
38
].
6.1
Example: SSU
Here we use an example fromMirarab et al. [
49
]. They provide four
datasets, M2, M3, M4, and 16S.B.ALL, for assessing the perfor-
mance of phylogenetic placement. The first three are simulated
datasets, which we used to assess the accuracy of alignments in
[
38
]. Here we use the last one, which is based on actual data. It
consists of a curated MSA of 13,822 bacterial SSU rRNA
sequences, taken from the Gutell Comparative Ribosomonal Web-
site (CRW) [
50
], and 13,821 fragmentary sequences, which are
originally included in the CRW alignment but ungapped and artifi-
cially truncated.
Suppose a situation where we already have an MSA (
exis-
tingmsa
) consisting of 13,822 sequences, which are manually
curated, and we have newly sequenced 13,821 fragments (
frags
)
in a metagenomics project. Both files,
existingmsa
and
frags
,
are in the multi-fasta format. To build a full alignment consisting of
27,643 sequences, use
rRNA
in which full DP is used for computing the distances between the
sequences in the existing MSA and new fragments. A faster option
based on the number of shared 6mers is also available.
The latter option is recommended unless the data is divergent.
If the new sequences were all from a single known species, this
is a standard problem of mapping short reads to the (genomic)