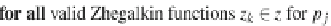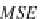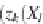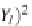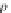Biomedical Engineering Reference
In-Depth Information
sigmoid representation (specified at runtime). Given a target gene
g
i
, our algorithm
determines a ranking of the best predictors for
g
i
with minimal error in the expected
output and the actual output
Y
i
(i.e. the best-fit Zhegalkin function on the input
X
i
),
over all the
i
1, 2,
...
,
m
gene expression state observations.
In line 2 of the algorithm, the method iterates through all possible predictor
combinations
p
j
=
p
for the target gene
x
i
. For each
p
j
, the method considers
all valid Zhegalkin functions
z
k
∈
∈
z
(line 3) by iterating over all possible coefficient
combinations corresponding to Boolean functions. In line 4, the algorithm determines
the mean squared error (MSE) of the expected output and actual output for each
Zhegalkin function
z
k
. The MSE is used to measure how well the Zhegalkin function
fits or matches the actual output expression values. In line 6, the minimum MSE
of all Zhegalkin functions for predictor
p
j
is chosen as representative MSE for
p
j
.
After the MSEs for all predictors have been determined, the predictors are sorted by
their MSE (line 8), and the ranked predictors (as well as their Zhegalkin functions)
and corresponding MSE are returned.
The algorithm returns a ranked list of predictors rather than a single predictor
with the lowest MSE for a several reasons. The main reason is as follows: The
actual or correct predictor is expected to be one the top ranked predictors, as the
actual predictor should best match the input data with a low MSE. However, the
“correct” predictor may not have the lowest MSE if the gene expression samples
are not adequately distributed. Ideally, the samples should be uniformly distributed
throughout the state space. However, a limited number of samples may result in
some areas in the state space being inadequately covered. For example, if none of
the samples were represented in a particular region, several Zhegalkin functions or
predictors may match equally well. The other reason is that the expression data may
be noisy or may contain errors, resulting in a higher MSE for the “correct” predictor,
potentially decreasing its rank. The ranked list ensures that the “correct” predictor is
not prematurely disqualified from the results.
In general, given adequately distributed expression samples, the top ranking
predictor can be selected as the inferred predictor, if the top ranked predictor has
significantly lower MSE than the second ranked predictor. In the next section, we
describe one method for selecting a predictor from the ranked list. Otherwise, if sev-
eral predictors have similarly low MSE (due to sample distribution or noisy data),
the ranked list can be used to help guide follow-up lab experiments to test and verify
those particular predictors (and corresponding gene regulation function).