Information Technology Reference
In-Depth Information
and yields the log-likelihood of these two nodes being matched. When u is an
insertion state, it takes as input at the sequence pro
le context of one node and
yields the log-likelihood of this node being an insertion. The sequence pro
le
context of node i is a 21
ð
2w
þ
1
Þ
matrix where w
¼
5
;
consisting of the mar-
ginal probability of 20 amino acids and gap at 2w
þ
1 nodes indexed by i
w
;
i
w
þ
1
;
...
;
i
;
i
þ
1
;
...
;
i
þ
w
:
In case that one column does not exist (when
i
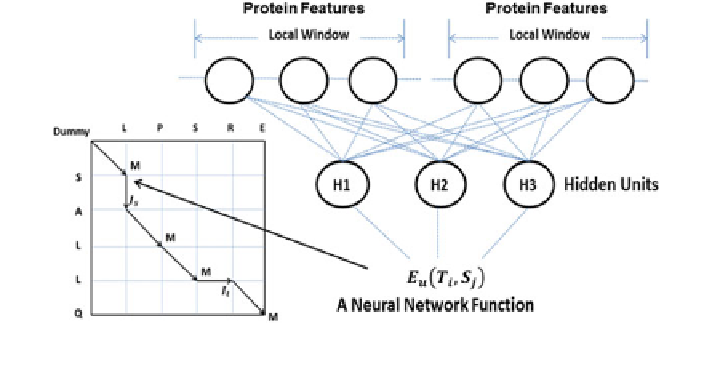
N), zero is used. See Fig.
2.4
for an illustration of how to use
neural networks to estimate local similarity of two MRF nodes.
We train the parameters in E
u
by maximizing the occurring probability of a set of
reference alignments, which are generated by a structure alignment tool DeepAlign
[
15
]. That is, we optimize the model parameters so that the structure alignment of
one training protein pair has the largest probability among all possible alignments.
Notice that by using neural networks the objective function of Eq. (
2.4
) is neither
concave nor log-concave, so it is challenging to
w for i
þ
w
[
find globally optimal solution. Here
we use the Limited memory BFGS (L-BFGS) algorithm to solve it to suboptimal.
To obtain a good solution, we run L-BFGS several times starting from different
initial solutions and return the best suboptimal solution. A L
2
-norm regularization
factor, which is determined by
fivefold cross validation, is used to restrict the search
space of model parameters to avoid over-
tting.
u
i
;
j
denote the local alignment potential of a vertex
h
ð
;
;
Þ
Let
i
j
u
in the alignment
u
i
;
j
path. We calculate
h
from E
u
as follows.
u
i
;
j
¼
h
E
u
T
i
;
S
j
Exp
ð
E
u
Þ
ð
2
:
5
Þ
where Exp
which depends only on the alignment
state but not any specific protein pair. It is used to offset the effect of the back-
ground, which is the log-likelihood yielded by E
u
for any randomly chosen node
ð
E
u
Þ
is the expected value of E
u
;
Fig. 2.4 An example neural network for calculating node alignment potential, in which there is
one hidden layer. The function takes features from both proteins as input and yields one log-
likelihood score
Search WWH ::

Custom Search