Information Technology Reference
In-Depth Information
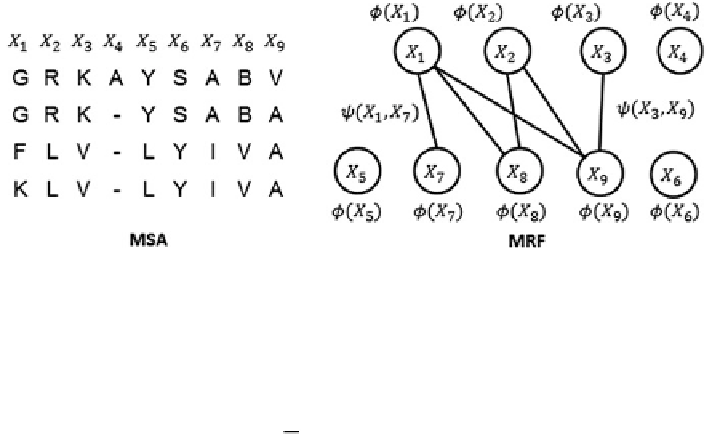
Fig. 2.1 Modeling a multiple sequence alignment (left) by Markov random fields (right)
that re
ects interaction between two nodes. Then, the probability of observing a
particular protein sequence X can be calculated as follows.
fl
1
Z
P
i
/ð
PX
ðÞ¼
X
i
ÞP
ð
i
;
k
Þ
wð
X
i
;
X
k
Þ;
ð
2
:
1
Þ
where Z is the normalization factor (i.e., partition function). If P X
is Gaussian,
this MRF is called Gaussian Graphical Model (GGM). In this case, Xi
i
shall be
interpreted as a 21-dimension binary vector, in which each element corresponds to
one amino acid type or gap.
We use two kinds of information in MRFs for their alignment. One is the
occurring probability of 20 amino acids and gap at each node (i.e., each column in
MSA), which can also be interpreted as the marginal probability at each node. The
other is the correlation between two nodes, which can be interpreted as interaction
strength of two MSA columns.
ðÞ
2.2 Estimating the Parameters of Markov Random Fields
In this section, we describe how to estimate the parameters in the
w
function of the
MRF. As stated above,
w
is a pairwise amino acid preference function for edge
ð
;
Þ
ecting interactions between two nodes (residues). A couple of strategies
can be used to estimate residue interaction strength. One is to estimate residue
co-evolution strength from the MSA and then use this co-evolution strength as
residue interaction strength. Mutual information (MI) is a simple and popular way
to estimate residue co-evolution strength. However, mutual information (MI) is
only a local statistics (i.e., the MI of 2 positions is calculated independent of the
other positions), so MI is not very accurate in estimating residue co-evolution
strength. Instead of using MI, we can use direct information (DI), which is a global
statistics (i.e., measuring the residue co-evolution strength of two positions con-
sidering other positions). DI can be calculated by some contact prediction programs
i
k
re
fl
Search WWH ::

Custom Search