Biomedical Engineering Reference
In-Depth Information
handling
complexes
that
undergo
significant
conformational
changes
on
binding.
170
HADDOCK is able to deduce native-like structures for protein complexes
by treating CSP data in a qualitative binary fashion, in which AIR restraints
are either present or absent. This suggested that the refinement process could
be enhanced by a more detailed interpretation of the dependence of chemical
shift data on torsion angle, electric field, ring current and hydrogen bonding
effects. The first implementation of this concept was provided by CamDock,
which starts from crystal or NMR structures of the unbound components, then
performs ab initio docking procedures to sample appropriate relative
orientations, followed by molecular simulation refinement driven by the
difference between experimental and SHIFTX-predicted chemical shifts, after
which the results are ranked with a composite energy score developed for the
CHESHIRE program.
171
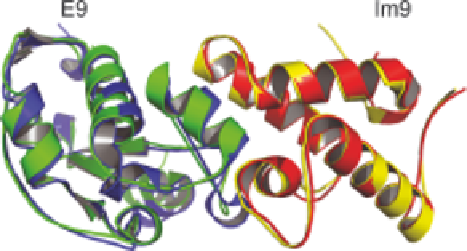
When applied to chemical shift changes for
1
H
a
,
13
C
a
,
13
C
b
and
15
N nuclei induced by the interaction between endonuclease
colicin E9 and its immunity partner protein Im9, the overall C
a
RMSD
between the CamDock model and the X-ray structure of the complex was
1.2
˚
(see Figure 3.9).
171
This example falls in the 'moderate' difficulty range
for flexible docking, with C
a
RMSD values for the unbound and bound
components between 0.9 and 2.0
˚
.
172
A further twist was introduced by Vendruscolo and colleagues, who
recognised that structures for one or more unbound partners might not always
be available. With data from experiments on the Ztaq/Anti-Ztaq affibody
complex, they deployed CHESHIRE to predict structures for the isolated
components from their chemical shifts in the bound state, which were then
fitted together by CamDock.
173
This problem lies in the 'difficult' range for
flexible docking, with unbound/bound C
a
RMSD values of 1.5-3.5
˚
,so
obtaining an overall C
a
RMSD of 1.1
˚
from the experimental solution
structure of the complex was impressive. Interestingly, the CamDock structure
of the complex was closer to the X-ray reference structure than CHESHIRE
Figure 3.9
Overlay of cartoons for structures of the complex between colicin
endonuclease E9 and immunity protein Im9, determined using X-ray
crystallography
(blue
and
red;
PDB
code:
1EMV)
and
using
CamDock
171
(green and yellow; PDB code: 2K5X).