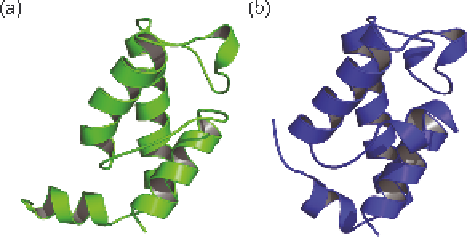Biomedical Engineering Reference
In-Depth Information
Figure 3.8
(a) Cartoon of the NOE-based structure of the second acyl carrier protein
domain from the deoxyerythronolide B synthase (DEBS ACP2), PDB
code: 2JU1. (b) Cartoon of a CHESHIRE chemical-shift-derived
structure for DEBS ACP2, with a backbone RMSD of 1.7
˚
to structure
2JU1.
157
assignments,
136
whereas CS-ROSETTA uses SPARTA and reads
1
H
N
,
1
H
a
,
13
C
a
,
13
C
b
,
13
C9 and
15
N data
137
), but in favourable cases both methods return
model structures with backbone RMSDs within 1.5
˚
of reference structures
determined by X-ray crystallography, for a wide range of protein topologies
(for example, see Figure 3.8). Although both approaches focus on backbone
chemical shifts, side-chain conformations in the models are also in good
agreement with reference structures, with RMSDs for all heavy atoms usually
,2.5
˚
; this probably reflects the quality of the description of hydrogen
bonding, torsion angles, side-chain packing and solvation in the refinement
phase.
137
CHESHIRE and CS-ROSETTA share the limitation of being computa-
tionally expensive, as both require thousands of CPU hours to run to
completion. Parallel processing can provide a useful degree of acceleration, but
a more radical approach is to take advantage of homology modelling, the
assembly of larger fragments by chemical shift threading and rapid genetic
algorithms for searching conformational space, as practiced by the CS23D
138
and GeNMR
140
servers. If a homologue of known structure with a sequence
identity .35% is present in the database, GeNMR is capable of reading the
sequence and shift assignments for a query protein and returning candidate
models with RMSDs y1.7
˚
from the reference structure in less than
20 min.
140
However, trials with designed proteins that share .95% sequence
identity but possess distinct three helix bundle and a/b-folds revealed that the
fragment selection algorithm of CS23D can sometimes be 'tricked' by the high
similarity of the sequence profile, causing the procedure to converge on an
incorrect
final
structure;
by
contrast,
the
small-fragment
focus
of
CS-
ROSETTA enabled it to home in on the correct fold.
141
More generally, extensive testing with a 110-residue protein confirmed that
CS-ROSETTA is a robust method, generating correct models even if chemical
shift assignments are randomly omitted, or even completely absent for certain