Biomedical Engineering Reference
In-Depth Information
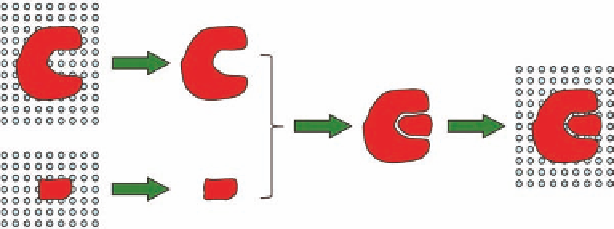
A
B
C
A
FIGURE 2.3
Simplii ed binding process. (A) Desolvation of protein and ligand (large unfavorable energy
change). (B) The binding process (small favorable energy change). (C) Solvation of the protein-ligand complex
(large favorable energy change).
provides an accurate structure, and accordingly such a structure is suitable for biostructure-based
drug design. Structures based on 3 Å resolution electron densities should be used with caution.
The stereochemical quality of a protein structure should be carefully evaluated prior to using
it for biostructure-based drug design. Planarity of peptide bonds, bond lengths, bond angles, and
torsion angles should not deviate from average values. The protein backbone geometry is usually
validated by a Ramachandran plot, a plot of the two variable torsion angles (phi and psi) (Figure
2.2B). Special attention should be paid to the part of the protein involved in direct interactions with
ligands, e.g., l exible loops and binding site residues.
Binding of a drug molecule to a protein is a complicated process which is often considered as
composed of a number of discrete steps in order to simplify the understanding of the process: (1)
the protein may contain a cavity where the drug may bind, but prior to binding the water molecules
occupying the cavity has to be removed. Similarly, the drug molecule may be surrounded by water
molecules that have to be removed before binding. These two desolvation processes are associated
with an unfavorable energy change (Figure 2.3A). (2) The next step is the actual binding between
the drug molecule and the protein. The drug molecule may change conformation in order to i t into
the binding site of the protein, and this conformational change requires energy. The protein may
also change conformation, but this is usually ignored. The binding process requires that the drug
molecule and protein are complementary not only with respect to shape, but also with respect to
molecular properties. Positive parts of the drug molecule will bind to negative parts of the binding
cavity and vice versa, and hydrogen-bond donors will bind to hydrogen-bond acceptors and vice
versa (Figure 2.3B). (3) Finally, the complex between the drug molecule and the protein has to be
surrounded by water molecules (solvated), and this process is associated with a favorable energy
change (Figure 2.3C). Thus, the actual binding energy is a relatively small difference (typically on
5-20 kcal/mol) between the processes representing large destabilizing vs. stabilizing forces (for further
details see Chapter 1).
2.2 ANTI-INFLUENZA DRUGS
One of the innovative examples on biostructure-based drug design was the discovery of the anti-
inl uenza drug zanamivir (Relenza). Together with the subsequent discovery of the orally active
anti-inl uenza agent oseltamivir (Tamil u) they now constitute a classical example on a successful
drug design story.
Two glycoproteins, namely hemagglutinin and neuraminidase, present on the surface of the virus
are important for the replication cycle of the virus. The enzyme neuraminidase, or sialidase, is a gly-
cohydrolase responsible for cleavage of the terminal sialic acid residues from carbohydrate moieties
on the surface of the host cells and the inl uenza virus envelope. This process facilitates the release
of newly formed virus from the surface of an infected cell. Inhibition of neuraminidase will leave