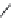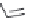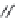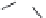Biomedical Engineering Reference
In-Depth Information
NH
NH
2
N
N
S
2
S
H
2
N
Dimaprit
Amthamine
NH
NH
N
N
S
S
H
H
H
H
HN
HN
(R)
N
N
N
N
Sopromidine
Impromidine
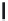
FIGURE 17.3
Reference histamine H
2
receptor agonists.
discovery of dimaprit (Figure 17.3), which was found during a search for H
2
receptor antago-
nists in a series of isothiourea derivatives. Dimaprit is an H
2
receptor agonist that is almost as
active as histamine at the H
2
receptor, but hardly displays any H
1
receptor agonism. Later it
was found that dimaprit is also a moderate H
3
receptor antagonist and a moderate H
4
receptor
agonist. Using dimaprit as a template, amthamine (2-amino-5-(2-aminoethyl)-4-methylthiazole)
was designed as a rigid dimaprit analog. Following the original suggestion of Green et al. that
the sulfur atom of dimaprit might act as a proton acceptor in a hydrogen bonding network with
the H
2
receptor (in analogy to the idea of the interaction of the imidazole ring with the receptor
protein), quantum chemical calculations by and synthesis of 2-aminothiazole analogs coni rmed
this idea. Amthamine combines a high H
2
receptor selectivity with a potency, which is slightly
higher compared to histamine, both
in vitro
and
in vivo
. An H
2
receptor agonist that is more
potent than histamine is the guanidine derivative impromidine. This ligand actually combines
a rather high H
2
receptor afi nity with a reduced efi cacy. Impromidine also shows moderate H
1
- and
potent H
3
-receptor antagonistic and H
4
-receptor agonistic activity. Interestingly, replacement
of the propyl-imidazole moiety of impromidine with an
-methyl-ethylimidazole group results
in the chiral analog (Figure 17.3). The
R
(−)-isomer, sopromidine, acts as a potent H
2
agonist,
whereas the
S
(+)-isomer is a weak H
2
antagonist. Both compounds posses only weak H
3
and H
4
antagonistic activities, making
R
(−)-sopromodine one of the most potent and selective H
2
agonist
to date.
α
17.3.3 H
2
R
ECEPTOR
A
NTAGONISTS
The identii cation of
N
α
-guanylhistamine as a partial H
2
agonist in a gastric acid secretion model
led to the development of the relatively weak H
2
antagonist burimamide (Scheme 17.2) following
the replacement of the strong basic guanidine group by the noncharged, polar thiourea, and side
chain elongation. Years later, it was shown that burimamide is also an H
3
and H
4
receptor partial
agonist. As H
2
receptor antagonist, burimamide lacked oral activity in man most likely due to its
moderate potency. Nevertheless, burimamide was the lead for the development of selective and
clinically useful H
2
receptor antagonists, such as cimetidine. Over time, many H
2
antagonists have
been described; almost all of them possess two planar
-electron systems connected by a l ex-
ible chain. The 4-methylimidazole moiety of cimetidine can easily be replaced by other heterocy-
clic groups. Replacement by a substituted furan (e.g., ranitidine) or thiazole ring (e.g., famotidine)
leads to compounds that are usually more potent at the H
2
receptor than cimetidine. Moreover, the
replacement of the imidazole moiety also eliminates the undesired inhibition of cytochrome P-450.
Most H
2
antagonists are rather polar compounds, which do not readily cross the blood-brain barrier.
π