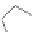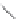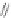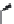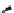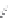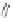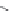Biomedical Engineering Reference
In-Depth Information
N
COOH
COOH
NH
2
COOH
NH
2
HOOC
HOOC
HOOC
NH
2
(2
R
,4
R
)-APDC (
15.93
)
LY354740 (
15.94
)
ABHxD-I (
15.95
)
HO
OH
O
N
COOH
HOOC
COOH
HN
COOH
COOH
COOH
N
O
N
HOOC
O
NH
2
O
NH
2
O
NH
2
NH
2
NH
2
Glu (
15.2
)
(1
S
,3
R
)-ACPD (
15.92
)
AMPA (
15.65
)
Ibotenic acid (
15.46
)
Quisqualic acid (
15.64
)
HO
O
OH
N
O
HN
N
COOH
COOH
COOH
COOH
N
O
HOOC
HOOC
O
COOH
O
NH
2
NH
2
NH
2
H
2
N
NH
2
(
S
)-2-Aminoadipic acid (
15.96
)
(1
S
,3
R
)-Homo-ACPD (
15.97
)
(
S
)-Homo-AMPA (
15.98
)
(
S
)-Hexyl-HIBO (
15.99
)
(
S
)-Homo-quis (
15.100
)
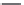
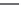
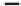
FIGURE 15.20
Structures of some mGluR agonists (upper row) and some Glu analogs acting at iGluRs and/
or mGluRs (middle row) and the corresponding homologs acting selectively at mGluRs (lower row).
Apart from Glu itself (1
S
,3
R
)-ACPC (
15.92
), ibotenic acid (
15.46
) and quisqualic acid (
15.64
)
were among the i rst potent metabotropic agonists, though fairly nonselective. Synthesis of homologs
of these and other Glu analogs afforded compounds with more selective activity at mGluRs. Thus,
(
S
)-aminoadipic acid (
15.96
) was shown to be a mGluR2 and mGluR6 agonist, (1
S
,3
R
)-homo-
ACPD (
15.97
) a Group I agonist, whereas (
S
)-homo-AMPA (
15.98
) showed specii c activity at
mGluR6, and no activity at neither iGluRs nor at other mGluRs. A number of HIBO analogs includ-
ing (
S
)-hexyl-HIBO (
15.99
) show group I antagonistic activity and (
S
)-homo-quis (
15.100
) is a
mixed group I antagonist/group II agonist. The effect of backbone extension of different Glu ana-
logs is often unpredictable, but chain length is nevertheless a factor of importance (Figure 15.20).
15.8.2 C
OMPETITIVE
M
ETABOTROPIC
G
LUTAMATE
R
ECEPTOR
A
NTAGONISTS
One of the i rst potent mGluR antagonists to be reported was (
S
)-4CPG (
15.101
), and it has been
used extensively as a template for designing further potent and selective antagonists at mGluR1. The
α
-methylated analog, (
S
)-M4CPG (
15.102
), is an antagonist at both mGluR1 and mGluR2. It has
been shown that the antagonist potency is increased by methylation at the 2-position of the phenyl
ring. Thus (+)-4C2MPG (
15.103
) is approximately i vefold more potent than the nonmethylated par-
ent compounds. It is notable that most 4-carboxyphenylglycines show selectivity for the mGluR1
subtype with no or weak activities at the closely related mGluR5 subtype. One exception to this rule
is (
S
)-hexyl-HIBO (
15.99
), which is equipotent as an antagonist at mGluR1 and mGluR5.
α
-Methylation has been widely used to derive antagonists from agonists. Maintaining the selec-
tivity proi les as of their parent compounds, MAP4 (
15.104
) and MCCGI (
15.105
) antagonize
mGluR2 and mGluR4, respectively, albeit with signii cantly reduced antagonist potency compared
to the parent agonist.
Subst it ut i ng agon ist s w it h bu l k y, l ip oph i l ic side cha i n s ha s b e en a much mor e suc c essf u l approa ch
to the design of potent antagonists. Two of the early compounds in this class are 4-substituted ana-
logs of Glu such as
15.107
and
15.108
, which are potent and specii c antagonists for mGluR2 and
mGluR3. Interestingly, compounds with small substituents in the same position, such as (2
S
,4
S
)-
Me-Glu (
15.106
), are more potent agonists at mGluR2 than Glu, with some activity at mGluR1
but without appreciable activity at mGluR4. Thus by increasing the bulk and lipophilicity at the
4-position to give such “l yswatter” substituents, the selectivity for group II is retained, and even
increased, but the compounds are converted from agonists to antagonists. One of the most potent