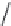Biomedical Engineering Reference
In-Depth Information
OH
OH
H
H
5α-Reductase
HH
HH
O
O
NADPH
NADP+
(A)
Testosterone
Dihydrote stosterone (DHT)
F
F
F
F
NH
O
O
NH
O
HN
F
O
F
NH
2
PADPR-N
H
H
H
HH
HH
HH
O
O
O
H
H
H
H
H
H
(C)
NADP-dihydroinasteride adduct
(B)
Finasteride
Dutasteride
FIGURE 11.4
(A) 5α-Reductase catalyzed conversion of testosterone (T) to dihydrotesterone (DHT);
(B) chemical structures for i nasteride and dutasteride; and (C) the proposed structure of the NADP-
dihydroi nasteride adduct. PADPR, phosphoadenosine diphosphoribose.
HMG-CoA to mevalonate, the rate-limiting step of the entire pathway. Inhibition of HMG-CoA
reductase provides a very attractive opportunity to inhibit cholesterol biosynthesis because no
buildup of potentially toxic precursors occurs upon inhibition. In 1976, Japanese microbiologist
Akira Endo isolated a series compounds including ML236B (compactin) from
Penicillium citrinum
with powerful inhibitory effect on HMG-CoA reductase. Since then, seven HMG-CoA reductase
inhibitors have become marketed drugs for lowering cholesterol levels (Figure 11.5).
These HMG-CoA reductase inhibitors, commonly referred to as statins, have accounted for
the majority of prescriptions for cholesterol-lowering drugs worldwide. All the statins in clinical
use are analogues of the substrate HMG-CoA with an HMG-like moiety, which may be present in
an inactive lactone form in the prodrugs (Figure 11.5.). These statins are classii ed in two groups
according to their molecular structures. Type I statins, including lovastatin and simvastatin, are
lactone prodrugs originally isolated from fungi. They are enzymatically hydrolyzed
in vivo
to
produce the active drug. Type II statins are all synthetic products with larger groups attached to
the HMG-like moiety. All the statins are competitive with respect to HMG-CoA and noncom-
petitive with respect to NADPH, a cosubstrate of the reaction. Crystal structures of HMG-CoA
reductase complexed with six different statins showed that the statins occupy the HMG-binding
region, but do not extend into the NADPH site. The orientation and bonding interactions of the
HMG-moiety of the statins resemble those of the substrate complex. However, from combina-
tion crystal structures, binding thermodynamics, and SAR studies it is clear that the 5
-hydroxyl
group of the acidic side-chain acts as a mimetic of the tetrahedral intermediate of the reduction
reaction. The multiple H-bonds between the C-5-OH of the statins and the HMG-CoA reductase
active site contribute signii cantly to the tight binding of the statin inhibitors. Strictly speaking,
the HMG-CoA reductase inhibitors are not products of rational design; rather they were identii ed
′