Biomedical Engineering Reference
In-Depth Information
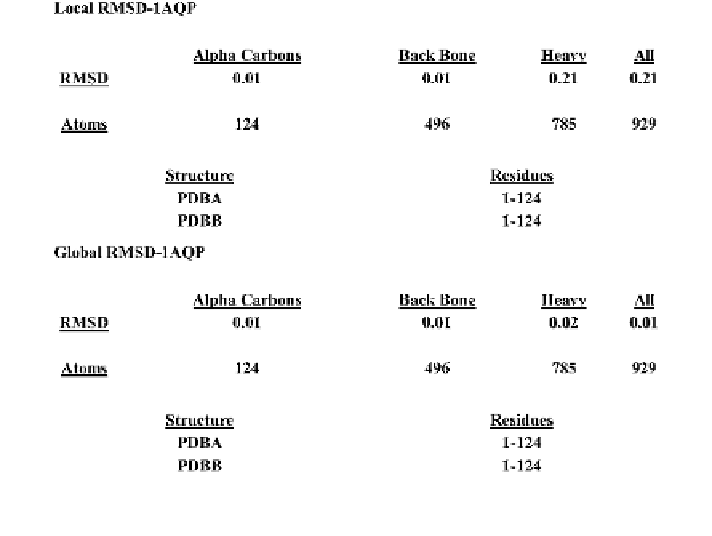
Fig. 14 The RMSD calculations from the superposition of the 1AQP with the minimized model
for 929 residues
Based on our alternative approach, we were able to achieve accurate models for
known proteins based on PDB files or amino acid sequences. The alignments were
shown between the modeled and known structures via Clustal-W. The alignment
differences given by the standard deviation (RMSD) showed minimal differences.
To summarize, the peptide braid provided the mathematical foundation that
described the model and the three dimensional structure. The union arc-lengths for
the model were given by the dipole moment (plane) of the N-H and the C=O. The
differential geometry was used to create the algorithm for the projections in the
rotations of the angles and the surface of the structure.
The minimizer calculated complex structures based the interactions between
each of the 20 types of residues. This only gives 210 possible interactions and
includes the interactions between the solute with the sodium or other charged ions.
The residue interaction was obtained by assigning properties to the surface charges
and hydrophobicity.
For the examples considered, we used Clustal-W and STRAPS for the align-
ment and RMSD for the verification. The simulations assumed residue interactions
but ignored charges and sulfur bridges that are not systematic (all high by an eV or
two). To make the model more physiological, we added the solute component, and
assumed that the structure was formed in plain water with no ionic charge, and a
size of *1.8 Å. As an example, if we add a charged item to the solute, this did
influence charge attraction by shielding the charged residues. If the charged spe-
cies interacted at the site of hydrogen or other ions, the ionic size would alter the
Search WWH ::

Custom Search