Biomedical Engineering Reference
In-Depth Information
For this evaluation we used sequenced reads obtained from the
DREAM project [35], more specifi cally from challenge #1 of the
DREAM6 competition. We downloaded the original FASTQ fi les (paired-
end reads) representing mRNA-seq data from human embryonic stem
cells from
http://www.the-dream-project.org/challenges/dream6-
alternative-splicing-challenge
.
The FASTQ fi les were aligned to the reference human genome (version
GRCh37, February 2009) using TopHat version 1.3.1 [36]. In total ~86
million reads were aligned and converted from BAM to BED format. In this
evaluation, we measured how both CPU time and memory scale with
increased input size. The task was to identify all pair-wise overlaps between
a 'test' genomic interval fi le and a 'reference' genomic interval fi le. The
former was obtained from the set of ~86 million sequenced reads using
re-sampling without replacement (re-sampling of 1, 2, 4, 8, 16, 32 and
64 million reads), and the latter contained all annotated transcript exons
from the ENSEMBL database [37], as well as all annotated repeat elements
from the UCSC Genome Browser [17], that is a total of ~6.4 million entries.
As demonstrated in Figure 8.9, GenomicTools improves greatly on time
performance (speed-up of up to ~3.8 compared to BEDTools and ~7.0
compared to the IRanges package of Bioconductor) if the inputs are sorted,
Time evaluation of the overlap operation between a
set of sequenced reads of variable size (1 through 64
million reads in logarithmic scale) and a reference set
comprising annotated exons and repeat elements
(~6.4 million entries). Using GenomicTools on sorted
input regions yields a speed-up of up to ~3.8
compared to BEDTools and ~7.0 compared to the
IRanges package of Bioconductor
Figure 8.9


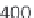


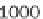
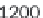
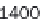

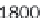


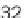
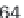
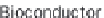

























































Search WWH ::

Custom Search