Database Reference
In-Depth Information
protein function and regulation. Quantitative “shotgun” proteomics has re-
cently emerged as a high-throughput technique for measuring the relative
abundances of thousands of proteins between two cellular conditions. In quan-
titative shotgun proteomics, proteolysis-derived peptides are measured with
liquid chromatography-tandem mass spectrometry (LC-MS/MS) and used as
surrogates of their parent proteins for relative quantification. By employing
stable isotope labeling, the proteomes under comparison are mixed together
and analyzed in one LC-MS/MS run. Each peptide in the mixture of two
isotopically labeled proteomes has two mass-different isotopic variants, the
light isotopologue (e.g.,
14
N) from one proteome and the heavy isotopologue
(e.g.,
15
N) from the other. Because sample handling in proteome measure-
ments is highly complex, proteome quantification requires rigorous statistical
approaches. Moreover, the size of the experimental data easily reaches sev-
eral gigabytes, thus challenging data analysis algorithms even further. Below,
we describe some of these challenges and approaches to address them for
metabolically labeled proteome quantification.
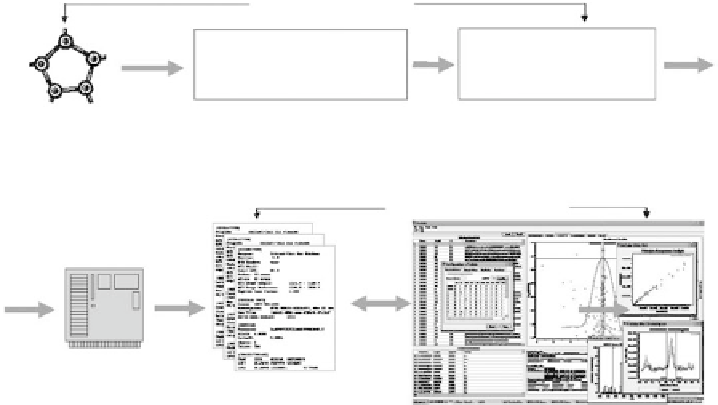
Figure 8.10 depicts the overall pipeline for proteome quantification from
data generation through data processing and analysis to visualization. The
resulting software system is called ProRata,
95
,
96
which is available as open
source from http://www.MSProRata.org. The analytical engine of ProRata
is available both as a serial C++ code and as a parallel
pR
code, and the
graphical user interface allows manual data interrogation for visualization and
Experimental Step
~3 GB raw data +
~50,000 MS, MS/MS files
~1 KByte each
Liquid chromatography-
Mass spectrometry (LC-MS)
24 hours measurements
Sample of ~2,000
labeled proteins (N
15
)
in different ratios
Quantification Step
Sequence Id Step
DBDigger+SEQUEST
~15-18 hours
ProRata: ~50,000
chromatogram files;
~1 KB each
ProRata: Analysis and visualization
Figure 8.10
Data processing pipeline in mass spectrometry proteomics for
proteome quantification.