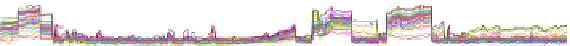Biology Reference
In-Depth Information
825 001
850 000
Genbank
annotation
yfmD
yfmC
yfmB
yflS
yflP
nosA
yfl
yflK yflH
mapB
yfkT
yfkQ
yflT
pel
citS
citM
nagP
yflB
yflA
citT
yflN
yflE
yfkR
Transcriptional
landscape
(+)
Annotated genes
and new transcription
segments
(+)
S257
S259
S263
S265
S269
S272
S258
Upshifts
(
+
)
(
−
)
Downshifts
and predicted TUs
Annotated genes
and new transcription
segments
(
−
)
S268
S271
Transcriptional
landscape
(
−
)
FIGURE 6.3
Bacillus subtilis transcriptional landscape determined by tiling arrays. A sample segment of
25 kbp of the chromosome is shown. The Genbank annotation is displayed above the
expression data collected in 269 hybridizations accounting for 104 different biological
conditions selected to maximize the diversity of lifestyles (
Nicolas et al., 2012
). For both
strands of the chromosome, 50 transcriptional profiles (coloured curves) illustrative of the
diversity observed in the total data set are drawn along with the new transcription segments
delimited (coloured boxes). The positions of signal upshifts and downshifts, also represented,
were shown to correspond mostly to transcription start sites and terminators and the
associated transcriptional units (TUs) were predicted.
transcription signal (in log2-scale) associated with a 95% credibility intervals that inte-
grate out probe affinity effects as measured fromgenomic DNA hybridization (
Nicolas
et al.
,2009
). The lower bound of credibility interval was then compared to a threshold
corresponding to 10
the chromosome median, used here as a proxy for the back-
ground. Of note, reverse transcription artefacts (limited by addition of actinomycin
D) cause well above background expression signals on the antisense strand that cannot
be identified by the aforementioned approaches. The authors found that visual inspec-
tion of transcriptional landscapes and quantitative measures of the reproducibility of
the signal could serve to filter out these artefacts.
Differential expression assessment plays a central role inmicroarray data analysis.
This is particularly true for experimental set-ups comparing a limited number of
biological conditions such that the research question can be easily addressed by estab-
lishing lists of differentially expressed genes. Hypothesis testing in linear models
represents the framework of choice for this purpose with well-established statistical
procedures (e.g. analysis of variance, ANOVA) that extend the popular Student
t
-test for comparing the mean of two samples, see, for instance,
Smyth (2005)
.
After log-transformation or more general variance stabilizing transformation
(
Huber
et al.
, 2002
), expression levels are written as a sum of terms representing