Biomedical Engineering Reference
In-Depth Information
1,000,000
100,000
10,000
1000
100
10
1
0
0.3 mg/kg
1 mg/kg
3 mg/kg
6 mg/kg
0
100
200
300
Time (min)
400
500
600
FIGURE 33.4
Semilogarithmic graphical representation of ame-
diplase plasma concentration versus time following single intra-
venous administrations at different doses in rats. Data is expressed
as mean (n
¼
8, 4 males
þ
4 females).
33.3.3.3 The
In Vivo
Fate of Amediplase in Rats The in
vivo fate of
125
I-amediplase has been studied in the rats by
intravenous administration of doses ranging from a trace
dose (50
m
g/kg body weight) to 1.0 mg/kg body weight.
Amediplase is relatively rapidly cleared from plasma,
mainly through hepatic route. Its kinetic profile is favorable
as compared to alteplase. At the used doses, liver uptake is
not saturated and is around 16-18%, as compared to 80% for
alteplase. Parenchyma liver cells and Kupffer cells are the
major contributors to hepatic uptake. Plasma clearance is
substantially reduced in hepatectomized rats. Amediplase
plasma clearance is marginally reduced in nephrectomized
rats indicating a minor role of kidney in its clearance
(Figure 33.5). Tissue distribution of amediplase, apart from
liver and kidneys, is limited to muscle, bones, and skin [21].
FIGURE 33.5
125
I-Amediplase was injected intravenously in
control rats (
), nephrectomized rats (
), or hepatectomized rats
(
&
). At the indicated time point, plasma samples were taken and the
radioactivity in the plasma was counted. Data are expressed as
percentage of the injected dose (mean
standard deviation, n
¼
4).
intravenous, paravenous, or intra-arterial route in the rabbit.
Amediplase is devoid of mutagenic properties. In the rat,
amediplase at 12.5, 25, and 50mg/kg from day 6 to day 15
postcoitus did not show teratogenic or embryofetal toxic
effect, while being maternotoxic at the highest dose. In the
rabbit, at 1, 3, and 10mg/Kg from day 6 to day 18 postcoitus
amediplase produced (similarly to tenececteplase) embry-
otoxic effects due to exaggerated pharmacology. In a battery
of eight safety pharmacology tests (CNS, cardiovascular,
gastrointestinal), amediplase did not reveal any unexpected
toxic effects.
33.3.4 Toxicology
Acute and subchronic toxicity studies in rat, dog, and
monkeys have not revealed any unexpected toxicity. Acute
toxicity studies in rats showed that the no-observed-adverse
effect level (NOAEL) is 10mg/kg. The acute NOAEL in
dogs was 1.0mg/kg and exaggerated pharmacological
effects were observed at 3 and 10 mg/kg. Subacute toxicity
in rats at 12.5, 25, and 50 mg/kg repeated for 14 days showed
that 12.5mg/kg/day is the NOAEL and toxic and/or phar-
macological effects were reported at the higher doses.
Subacute toxicity in dogs at 1 and 2 mg/kg repeated for at
least 9 days showed that 2mg/kg can be considered the
NOAEL and longer treatment time were prevented by
immunological response against a human protein. Subacute
toxicity in monkeys at 0.5, 1, 2, and 4mg/kg repeated for
14 days showed that 0.5mg/kg/day is apparently the
NOAEL and pharmacological effects were reported at
the higher doses. Amediplase clinical formulation showed
adequate local tolerance following single administration via
33.4 HUMAN STUDIES
33.4.1 1K2 Study: A Phase I Single Ascending
Dose Study
The clinical development of amediplase started with the first
administration to man in a Phase I single ascending dose
study (coded 1K2) [22].
Twenty healthy male volunteers completed a two-session,
double blind, placebo-controlled, crossover study, to eval-
uate the tolerability, pharmacokinetics, and pharmaco-
dynamics of amediplase. Patients received placebo and
one out of the following intravenous dose of amediplase,
each separated by a 3-day wash out period: 0.1mg/kg
(n
¼
2), 0.25 mg/kg (n
¼
6), 0.5mg/kg (n
¼
6) over 10 min
and 0.25 mg/kg (n
3) over a 5-10-
min period. The treatment was well tolerated. No clinically
¼
3) and 0.5mg/kg (n
¼




















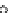




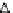




















Search WWH ::

Custom Search