Biomedical Engineering Reference
In-Depth Information
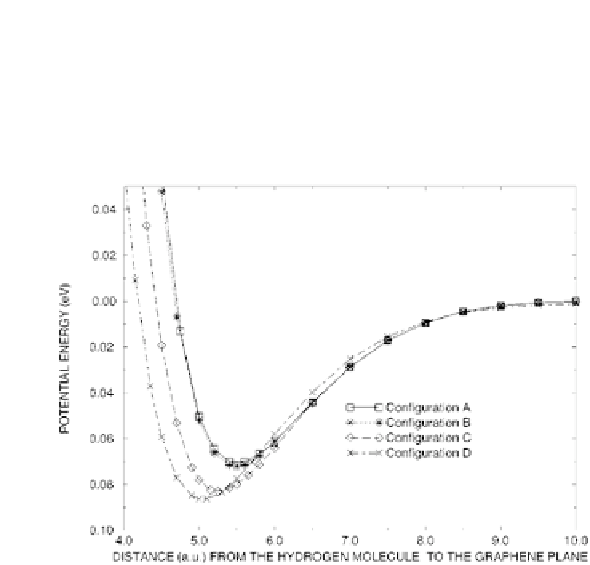
The interaction of an H
molecule with a graphene sheet has been
studied with LDA-DFT calculations [3] varying both the molecule
orientation and the adsorption site. The obtained energy curve,
reported in Fig. 8.4, show the typical van der Waals behavior for
physisorption with the different orientations and adsorption sites.
2
Figure 8.4
H
potential energy of nearby a graphene layer. Different
configurations of the molecule have been considered with the
axis perpendicular (A, B, C) or parallel (D) to the graphene
layer: above a carbon atom (A), above the center of a C-C bond
(B), above the center of a hexagon (C), above the center of a
hexagon (D). From Ref. [3].
2
Hydrogen uptake in GNFs has been simulated with conventional
LJ potentials showing that significant adsorption occurs only if the
interplanar distance is larger than 0.7 nm remaining, in any case,
below 1.5 wt.% at
= 10 MPa and RT [41]. With more accurate
potential parameters [102] the adsorption capacity of GNFs is about
2 wt.% at cryogenic temperature and ambient pressure. In the light of
the above results, recent experimental data reporting exceptionally
good adsorption excess are presumably affected by contaminants
such as oxygen or residual particles of metal catalysts used during
the synthesis [14, 43].
Therefore some authors have suggested that also graphene sheets
should be doped with alkali metals, such as Li and K, to increase the
uptake and some promising theoretical results have been published,
with special mention to Li doped graphene [15, 123].
P