Biomedical Engineering Reference
In-Depth Information
[1-4] have predicted the metal atoms tendency to attach onto
graphene through the hexagonal C ring centers, which are commonly
called the hollow sites (Fig. 5.1), with binding energies ranging from
about 0.1 to 2.0 eV. It is shown that the strong changes in carbon
sheet's properties can be indeed induced by such attachments to
graphene, the extent being dependent on the bonding mechanisms
involved.
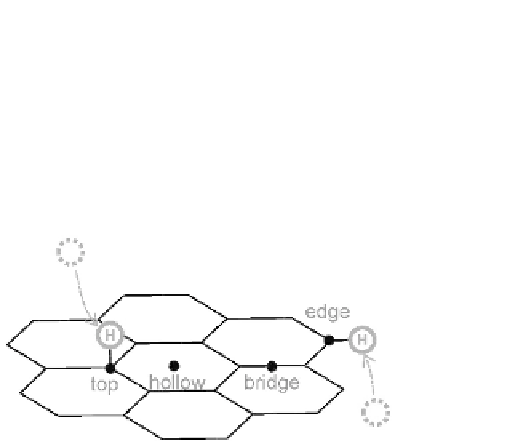
Figure 5.1
High-symmetry adsites on the graphene surface: top (nearest
surface atom coordination number = 1), hollow (=6), and
bridge (=2). Possible attachment through the edge defect of
nanostructured graphene is also shown.
In case of hydrogen, an atom approaching graphene preferen-
tially ends up at top sites (Fig. 5.1), i.e., H prefers a single-
coordination, and with appropriate time for relax, it reaches a very
stable chemisorbed state, albeit, going through an energy barrier of
about 0.2 eV [5]. This barrier arises from the need to disrupt C-C
bonds (which raises the system energy) in the process of creating
a stable C-H bond (which lowers the system energy). Reported H
atom adsorption energies vary slightly across the computational
studies, in the range from −0.6 to −0.8 eV with a negative sign
denoting exothermicity of the adsorption. The values primarily
depend on how the isolated H atom is detained on graphene. The
chemisorption is associated with the receiving carbon atom being
pulled out by about 0.33 Å from the initial planar geometry of
graphene. However, reconstruction due to the adsorption of a single
H atom is not limited by the receiving C atom, as clearly described
in Fig. 5.2a, which plots the displacement of C atoms from the
original graphene plane along the
-axis, the direction normal to the
graphene plane. Complementing it, Fig. 5.2b shows a local planar
distortion that is the distance of a C atom moving out of the plane
determined by its three nearest neighboring C atoms.
z