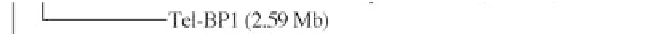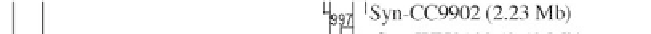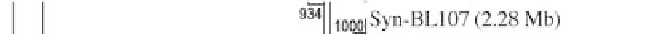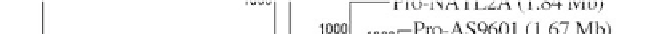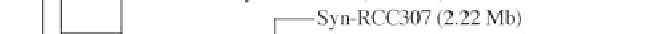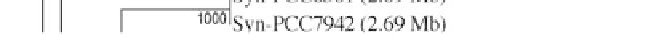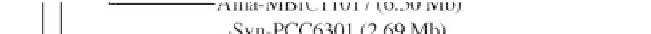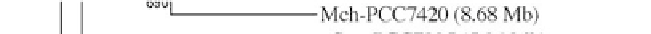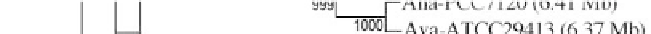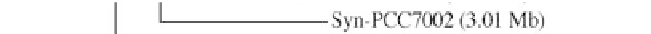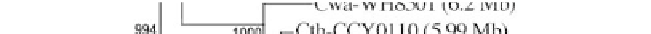Biology Reference
In-Depth Information
Figure 1:
Phylogenetic maximum likelihood (ML) tree based on the concatenated 23S-16S rDNA sequences of diverse
cyanobacterial lineages
taken from public databases. The scale bar represents the average number of nucleotide substitutions
per site. Genome sizes in megabases (Mb) are mentioned in parentheses. Trees were constructed using three methods (ML,
Neighbor Joining and Maximum Parsimony). ML bootstrap values are indicated only if the bootstrap values obtained with
the three methods are > 500 (1000 resamplings). See Table 1 for strain designations. With the kind permission of N. Tandeau
de Marsac,
Institut Pasteur, Unité des Cyanobactéries; CNRS, URA2172, F-75015, Paris, France.
[Frangeul
et al.
(2008)
BMC Genomics
9:
274; doi:10.1186/1471-2164-9-274]