Biology Reference
In-Depth Information
homologues have multiple functions are to identify highest hit and top hits. The uncharacterized
gene is assigned the function of the gene that is identifi ed as the highest hit by a similarity search
programme. Top 10+ hits are identifi ed for the uncharacterized gene. Depending on the degree of
consensus of the top hits, the query sequence is assigned a specifi c function.
Evolutionary trends are represented in the form of a phylogenetic tree. It is a graphical
representation of the evolutionary history of genes or species. The branching pattern of a tree
(topology) displays the evolutionary relationships of the strains. A tree consists of nodes and edges.
The nodes correspond to organisms and the edges show their relationships. The terminal nodes
possess only one linking edge and correspond to organisms for which we have data. These are
commonly designated as operational taxonomic units (OTUs). Internal nodes possess three linking
edges and correspond to hypothetical ancestor of one set of organisms. One unique internal node
with only two edges represents the root of the tree, the common ancestor to all taxa.
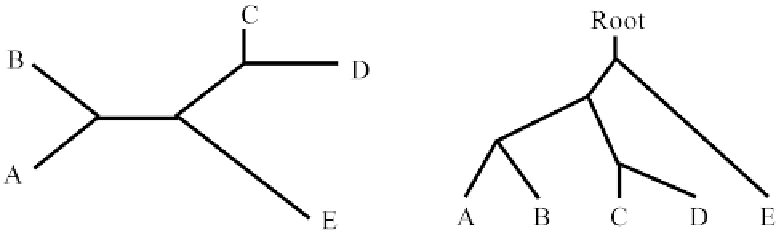
The phylogenetic trees may be represented as rooted or unrooted. Rooted or unrooted trees are
identical except that in the former nodes are present while in the latter nodes are absent but possess
only two connecting edges (Fig. 12). All proposed tree-building methods fall into two general classes:
algorithmic and optimal criteria approaches. In the former, a unique tree can be built by a series of
steps while in the latter all possible trees are examined and one best meeting the certain criteria is
chosen. In the algorithmic approach, the algorithm plays a fundamental role but in optimal criteria
approach the algorithm simply is a tool to evaluate the criteria. Algorithmic approaches are known
as distance methods because they compute a tree via a distance matrix composed of the distances
between each pair of sequences. A distance based algorithm is basically a procedure for building a
tree based on the matrix. If the outputted tree is to be additive it must obey four basic rules, i.e. (i)
all distances are positive; (ii) a distance between two points can be zero only when the two points
are actually the same; (iii) the distances are symmetrical and (iv) there are no shortcuts in the tree,
i.e. the distance between a-c cannot be longer than the sum distances of a-b and b-c. In addition, a
tree point condition is applied that is distance a-b cannot be larger than their distance to a third point
(the maximum distances a-c and b-c). Unweighted Pair Group Method (UPGMA) is an ultrametric
tree-building algorithm that follows all the rules mentioned earlier. UPGMA proceeds by inferring
one ancestral sequence per step. In the fi rst round UPGMA selects the least distant pair of sequences
(or one of them), summarizes their distance as the fi rst branches of a new tree and recalculates the
entire matrix with the pair as one entity (taking the mean distances). After N-1 steps (where N is the
number of sequences) the matrix is reduced to just one element. The last inferred ancestor is taken as
the root of the tree. Distance methods such as neighbor joining (NJ) use pair-wise distances (i.e. the
number of base differences between two sequences), calculated from aligned sequences and usually
Figure 12:
Representation of unrooted (left) and rooted (right) phylogenetic trees.