Biomedical Engineering Reference
In-Depth Information
Hpl
ot
M
p
l
ot
1.0
1.0
0.8
0.8
0.6
0.6
0.4
0.4
0.2
0.2
0.0
0.0
0
10
20
30
40
50
60
70
0
10
20
30
40
50
60
70
Nucleotide Position
Nucleotide Position
Ala
, derived with the common strategy (under the as-
sumption of
min
hel
=2
and
min
HL
=3
),whereweusedsamplesize
100
,
000
,
10
,
000
and
1
,
000
for
W
exact
=
−
1
(no window, thick gray lines),
W
exact
=30
(moderate window, thick
dotted darker gray lines) and
W
exact
=+
∞
Fig. 7.
Sampling results for
E.coli
tRNA
(complete window, thin black lines), respectively
Hpl
ot
M
p
l
ot
1.0
1.0
0.8
0.8
0.6
0.6
0.4
0.4
0.2
0.2
0.0
0.0
0
10
20
30
40
50
60
70
0
10
20
30
40
50
60
70
Nucleotide Position
Nucleotide Position
Fig. 8.
Sampling results corresponding to those of Fig. 7, obtained by employing the alternative
sampling strategy
sampling results shown in Fig. 7 indicate that for the common sampling strategy, con-
sidering a window of constant size
W
exact
(chosen to cover the size of hairpin-loops)
with a mixed preprocessing variant, actually yields a slight improvement of the result-
ing sampling quality, where the same time requirements are needed for generating the
respective sample sets.
Contrary to this observation, Fig. 8 demonstrates that when employing our alterna-
tive sampling strategy, the corresponding results are not significantly different for the
completely approximate preprocessing variant and for a mixed version on the basis of
a constant value for
W
exact
. Thus, to our surprise it does not matter if we consider a
constant window for exact calculations or simply approximate all inside and outside
values, which is not only an interesting observation itself, but also fortunately prevents
us from having to deal with an undesirable trade-off between reducing the worst-case
time complexity (by a linear factor) and sacrificing less of the resulting sampling qual-
ity. In fact, this means we may (without resulting significant quality losses) always use
the more efficient approximative preprocessing variant in order to reduce the worst-case
time complexity of the overall sampling algorithm.
However, all profiles perfectly demonstrate that due to the noisy ensemble distribu-
tion caused by approximating the highly relevant sequence-dependent emission
probabilities, the resulting sample sets usually contain many foldings that are rather
unlikely according to the exact distribution for the considered input sequence. For this
reason, it can not be recommended to employ one of the following otherwise rea-
sonable construction schemes for deriving predictions according to the entire sample
set: we should rather neither predict
γ
-MEA nor
γ
-centroid structures of the generated








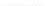

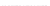




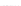

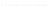













































































































































































































































Search WWH ::

Custom Search