Biomedical Engineering Reference
In-Depth Information
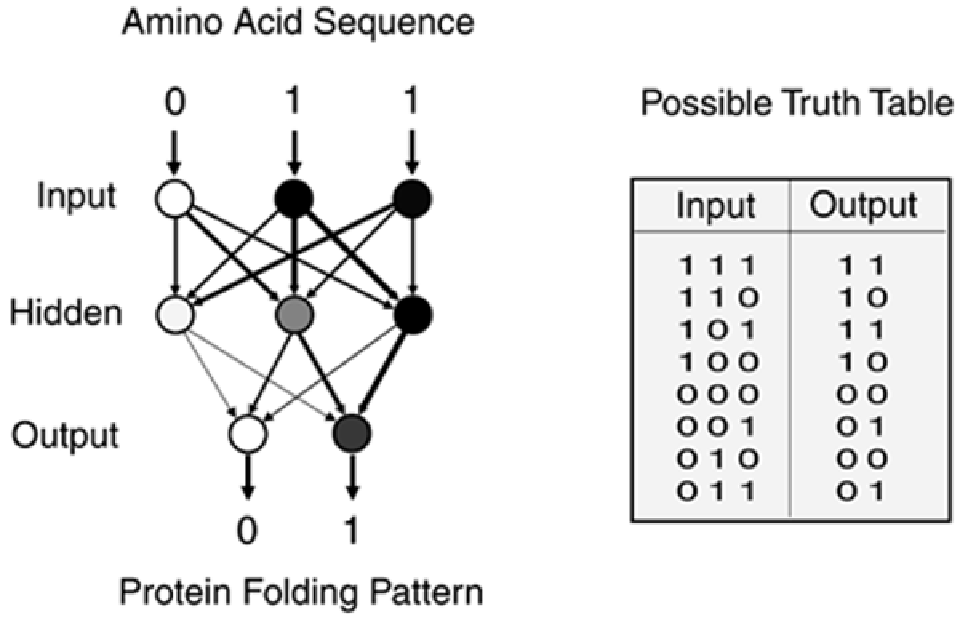
Artificial neural networks and other forms of machine learning are only one form of pattern matching
that have application in bioinformatics. Other approaches to pattern matching include techniques
such as genetic algorithms, which work by identifying the best fit for a function that is used to select
future generations. Hybrid systems of artificial neural networks supplemented by genetic algorithms
rule-based expert systems, and conventional, algorithmic programming hold particular promise in
bioinformatics.
In addition to simulating neural networks, the numerical processing capabilities of a computer are
commonly used to simulate the interactions of various proteins and drugs at their active sites. Data-
mining applications include searching patterns of known gene structures for newly discovered
patterns. Search engines are similarly instrumental to uncovering patterns or key works in local or
online databases. Pairwise sequence comparison, based on either BLAST or Smith-Waterman
dynamic programming techniques, form the basis for most sequence alignment operations. Using
online tools based on BLAST, it's possible for anyone with a connection to the Internet to evaluate all
possible ways of aligning one sequence against another in a reasonable time, even though the
number of such possible alignments grows exponentially with the length of the two sequences.
Statistical analysis is an important component of searching and pattern matching, especially in
dealing with uncertainty. For example, in the multiple sequence alignment problem in
Figure 1-15
,
statistical methods can be used to determine the best alignment of the four polypeptide strings
consistent with an alignment score that rewards perfect matches, and penalizes for imperfect
matches and the number and length of the gaps introduced in the final sequence. Non-statistical
methods of multiple sequence alignment can be used as well.
Figure 1-15. Multiple Sequence Alignment Problem. The unaligned
polypeptide sequences are shown at the top of the figure, and the resultant
sequence with gaps is shown at the bottom. Statistical and non-statistical
methods can be used to identify the optimum position of gaps and the
relative location of the high-scoring polypeptide sequences, as a starting
point for evolutionary modeling, for example.