Biomedical Engineering Reference
In-Depth Information
Pure ab initio methods of determining protein structure are based on sequence data and the physics
of molecular dynamics. Newtonian physics, atomic-level forces, and solving equations for the most
stable (minimum free energy) conformation or structure form the basis for these methods. Reasoning
from first principles assumes that the shape of a protein can be defined as a function of the amino
acid sequence, the temperature, pressure, pH, and other local conditions without knowledge of the
biology associated with the molecule. For example, the fact that a protein unfolds or becomes
denatured at elevated temperatures and reverts to its normal, active, folded state can be modeled
irrespective of the structure or function of the protein. However, unlike our knowledge of physics or
other hard sciences, our understanding of the first principles of molecular biology is largely
incomplete. As a result, attempts thus far at using first principles as the basis for determining protein
structure have been successful primarily as a means of defining limited areas (finishing) of the global
protein architecture. For example, with the overall protein structure approximately known, reasoning
from first principles can be used to define a particular bend in the structure.
Because of the computational demands associated with ab initio methods, assumptions and
simplifications are required for all but the smallest proteins. For example, just as the models of
individual neurons discussed earlier are composed of simple equations, instead of considering the
dozens of variables affecting each atom and bond in a real neuron, a common simplifying assumption
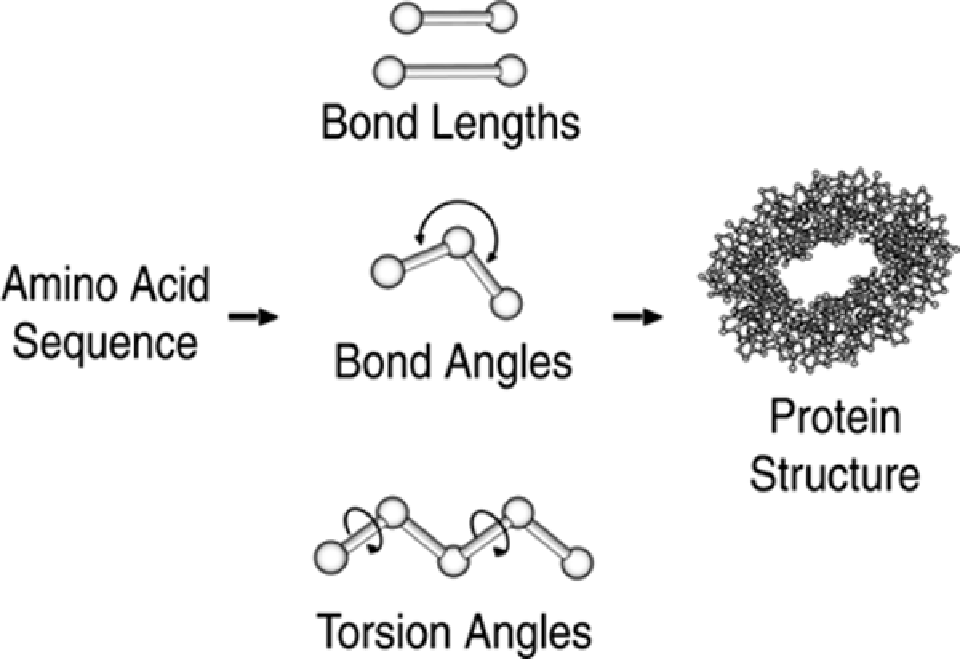
is that protein structure can be computed from bond lengths, bond angles, and torsion (dihedral)
angles (see
Figure 9-12
).
Figure 9-12. Ab Initio Protein Structure Determination. Based on the
protein's amino acid sequence (primary structure), secondary and tertiary
structures are computed. Tertiary structures typically takes the form of
xyz
coordinates for each atom in the protein molecule. Many ab initio methods
assume that protein secondary and tertiary structures are a function of
bond lengths, bond angles, and torsion angles.
Additional assumptions are that bond lengths are constant, and that bond length is a function of the
two atoms involved in the bond. Bond angles, which are a function of the relative position of three
atoms, are also assumed to be constant. Bond angles, which are limited to the range of about 100 to