Biomedical Engineering Reference
In-Depth Information
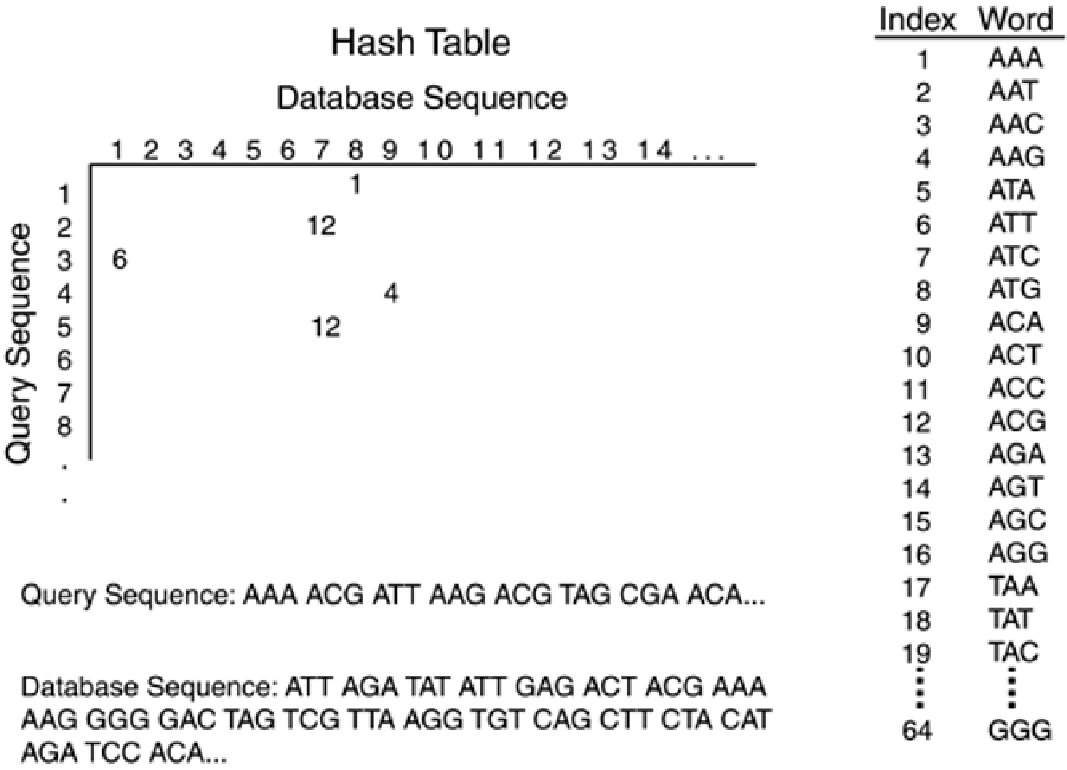
Next, the characters are compared to those in the database, which has previously been processed
into words of the same length. FASTA uses the Blosum50 substitution matrix to score the top-10
alignments (without gaps) that contain the most similar words. These words are then merged into a
gapped alignment, which is scored, producing an "optimized score." FASTA produces an expectation
score,
E
, which represents the expected number of random alignments with z-scores greater than or
equal to the value observed, thereby providing an estimate of the statistical significance of the
results.
Although FASTA was the first widely used program for sequence alignment against genome-length
sequences, and is still actively supported in both Web and workstation versions, BLAST is by far the
more popular of the word-based algorithms for sequence alignment. Like FASTA, BLAST is a heuristic
approach to sequence alignment that provides speed through a hashing technique. BLAST also differs
from FASTA in that words are typically 3 characters long for proteins and 11 characters in length for
nucleotide sequences.
Like FASTA, BLAST also searches a pre-computed hash table of sequences in the protein or DNA
database. However, where BLAST excels is that the matching words are then extended to the
maximum length possible, as indicated by an alignment score. The top-scoring alignments in a
sequence, called maximal-scoring pairs (MSPs), are combined if possible into local alignments. The
latest version of BLAST can attempt gapped alignment. However, this tends to extend computational
time significantly, compared to ungapped alignments. One of the major issues of both BLAST and
FASTA results is how to interpret the significance of results. An individual score depends on a number
of variables, including the lengths of the sequences being aligned, the gap penalties, and the
alignment scoring system used.