Biomedical Engineering Reference
In-Depth Information
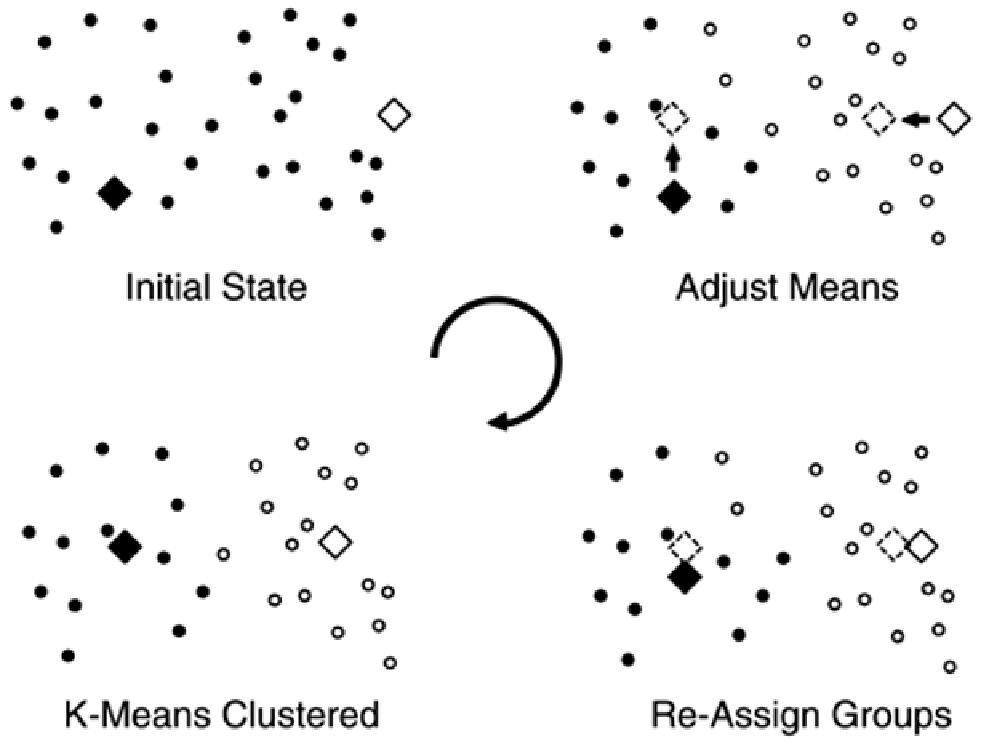
K-means clustering involves generating cluster centers (squares in
Figure 6-23
) in n-dimensions and
computing the distance of each data point to each of the cluster centers. Data points are assigned to
the closest cluster center. A new cluster position is then computed by averaging the data points
assigned to cluster center. The process is repeated until the positions of the cluster centers stabilize.
Clustering microarray gene expression data is useful because it may provide insight into gene
function. For example, if two genes are expressed in the same way, they may be functionally related.
In addition, if a gene's function is unknown, but it is clustered with genes of known function, the
gene may share functionality with the genes of known function. Similarly, if the activity of genes in
one cluster consistently precedes activity in a second cluster, the genes in the two may be
functionally related. For example, genes in the first cluster may regulate activity of genes in the
second cluster.
Common classification methods applied to gene expression data include the use of linear models,
logistic regression, Bayes' Theorem, decision trees, and support vector machines. For example,
consider using Bayes' Theorem to classify microarray data into one of two groups, illustrated
graphically in
Figure 6-24
.
Figure 6-24. Bayes' Theorem Example. The data points A, B, and C can be
classified using Bayes' Theorem.