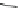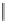Biomedical Engineering Reference
In-Depth Information
1,4-dihydroindeno [1,2-
c
]pyrazole and Phe656. The substitution of the methylene
bridge with a carbonyl group reduced the hERG affinity, without affecting the KDR
inhibitions.
In a recent study, Levoin et al. [
117
] combined homology modeling and docking
to refine a homology model of the H
3
receptor. The model of the H
3
receptor was
optimized in three different ways: ten independent molecular dynamics simulations
of the proteins embedded in the membrane; ten independent simulated annealing
(SA) runs with the most active compound of ten clusters obtained from the training
set of inhibitors of each protein; iterative simulated annealing (ISA) with a rigid
potent compound. The ligands were docked in the refined models and subsequently
the first ranked pose of each ligand was selected, and the affinity with the protein
was assessed using the DOCK_SCORE, Ligscore1-2, PLP1-2, Jain, PMF, and
Ludi_1-3 scoring functions. The performance of the prediction was then evaluated
using the ROC curve and the area under the curve (AUC). The best performing
model in the screening of the training set compounds was the one refined via the
ISA method, which achieved an AUC value of 0.73. Similar results were obtained
with the test set, where the model showed an AUC of 0.71. To predict also the
interactions with antitarget proteins, the same procedure was applied to refine the
homology models of hERG and CYP2D6. Also in these cases, the refinement
process leads to a better performance of the homology models by producing an
AUC
0.69 for both antitarget proteins.
Recently, Shamovsky et al. [
38
] used a combination of pharmacophore
modeling, QSAR and docking to develop a successful lead optimization strategy
that overcomes the undesirable interactions with hERG. The aim of the docking
experiments performed on a homology model of the hERG channel in the closed
state was to explain the intrinsic hERG binding. To achieve this aim two
compounds, which represent extreme cases of intrinsic hERG binding were docked
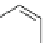
into the homology model of hERG. The compound A (Fig.
4
) makes cooperative
hydrogen-bond interactions with Ser624, whereas the compound B forms coopera-
tive
>
-stacking interactions with Tyr652. The docking poses suggest that also the
amino acids Leu622, Ser649, and Phe656 are involved in the intrinsic interactions
between the hERG channel and the blockers.
p
O
OH
N
Cl
O
Cl
O
N
N
OH
O
O
N
O
O
OH
Compound A
Compound B
Fig. 4 2D structure of the two compounds docked in hERG channel