Biology Reference
In-Depth Information
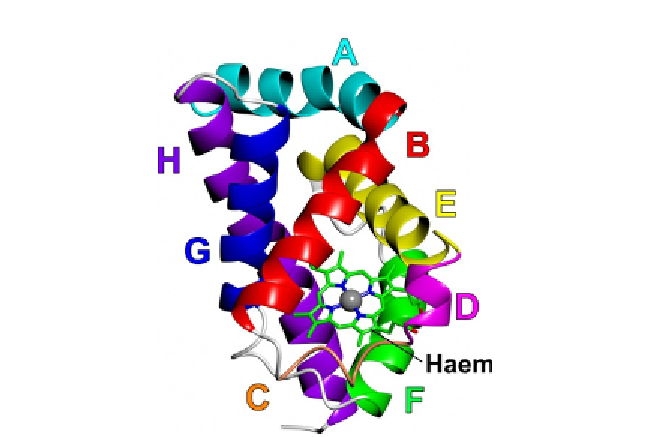
Figure 4.1 Backbone topology of Campylobacter globin, Cgb. The figure depicts the
3-over-3 a-helical fold of Cgb with the haem cofactor (PDB id¼2WY4;
Shepherd
et al., 2010
). Helices/regions are labelled according to conventional globin
nomenclature.
The structure of cyanide-bound Cgb was solved by X-ray crystallography
to a resolution of 1.35
˚
(
Shepherd et al., 2010
) andwas found to adopt a classic
3-over-3
-helical globin fold (
Fig. 4.1
). The helices constituting the globin
fold are labelled A-H in sequence order, according to standard globin nomen-
clature, and the amino acids within each helix are also numbered sequentially.
TheC andD regions adopt 3
10
-and
a
-helical conformations, respectively, and
the ligand-binding (distal) pockets of Cgb are constructed from the B-, E- and
part of the G-helices (
Fig. 4.1
). Structural overlays (
Fig. 4.2
) indicate consid-
erable structural homologywithVgb (RMSD
a
1.30
˚
, 110 residues), the glo-
¼
1.64
˚
, 134 residues) and sperm whale
bin domain of Hmp (RMSD
¼
1.83
˚
, 116 residues). Whereas Vgb is a dimer
(
Tarricone et al., 1997
) andHmp has an FAD-binding reductase domain (
Ilari,
Bonamore, et al., 2002
), Cgb was purified and crystallised as a monomer with a
single globin domain.
The identity of the amino acids in the B10 and E7 positions (i.e. the 10th
residue on the B-helix and the 7th residue on the E-helix) is known to be
important for modulating ligand binding. In mammalian globins, the E7
position is almost invariably occupied by a histidine. The HisE7 of Mb
myoglobin (swMb) (RMSD
¼