Environmental Engineering Reference
In-Depth Information
As can be guessed from the linear dependence on the electrode potential U in the
relations above, the thermochemical approach for handling the potential and pH
described above is in fact totally analogous to the method used in the classic
Pourbaix diagram. Figure 3.3 corresponds directly to reading of the redox potentials
in a Pourbaix diagram at the pH ¼ 0 line. The main difference is that the ab initio
binding energies can be obtained for all intermediates, i.e., also the species for reac-
tions and adsorbates not stable in the electrochemical environment. Whereas
Pourbaix diagrams were originally made for bulk transitions, DFT can provide the
numbers related to the surface adsorption, which are actually more relevant for elec-
trocatalytic reactions. In Fig. 3.4, we compare the first oxidation of the surface
obtained with ab initio DFT with the classic Pourbaix bulk values. As can be seen,
the results are very similar; the main difference is a downward shift in potential
for the surface oxidation compared with the bulk oxidation, which is what could be
expected. The computational Pourbaix diagrams can include all other surfaces and
dissolution potentials as well; Fig. 3.4 is just a simple example of what is possible
with this technique.
These results show that the electrochemical measurements can, via ab initio simu-
lations, be linked to phenomena at the atomic level, such as structural and electronic
effects and, in this case, binding energies on the surfaces.
Figure 3.4 Pourbaix diagram for the first oxidation step of Ni, Pt, and Au for the (111) surface
(dashed lines) [Nørskov et al., 2004] and the bulk (solid lines) [Pourbaix, 1966]. The oxidation
reactions corresponding to the solid lines are indicated to the right in the figure.










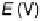



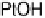




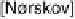


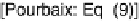































































Search WWH ::

Custom Search