Environmental Engineering Reference
In-Depth Information
The electronic contribution to the energy is obtained by integrating over all occu-
pied states. To a good approximation, the Fermi - Dirac distribution can be replaced by
a step function, and the integral can be performed up to the Fermi level:
E
elec
¼
ð
1r(1)f (1) d1
E
F
1
1r(1) d1
(2
:
8)
This integral diverges—a consequence of the wide band approximation—however,
this poses no problem. The relevant quantities are the differences in energy between
various states. It is natural to take the initial state as a reference. This gives
[Schmickler, 1986]
2p
ln
1
a
(q)
2
þ
D
2
E(q)
¼
1
a
(q)kn
a
(q)l
þ
1
2
h
vq
2
þ
D
(2
:
9)
1
a
(0)
2
þ
D
2
The first term is just the electronic energy of the reactant, including the interaction with
the solvent; the second is the energy of the solvent. The sum of these two terms is the
adiabatic part of the potential energy in the original Marcus and Hush theory. The last
part is the correction due to the finite width of the density of states; the denominator in
the logarithmic term is often left out, because it just adds a constant.
Figure 2.4 shows several potential energy curves for various values of the inter-
action strength D. With increasing interaction strength D, the energy of activation is
reduced. This effect, which corresponds to a catalytic action of the electrode, becomes
noticeable when D is of the same order of magnitude as the reorganization energy l.
Figure 2.4 Adiabatic potential energy E(q) for various values of D at equilibrium. The solvent
coordinate q has been normalized so that the initial state corresponds to q ¼ 0 and the final state
to q ¼ 1. The reorganization energy was taken as l¼ 1eV.
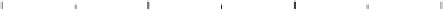






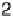
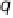
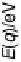

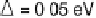

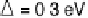






















Search WWH ::

Custom Search