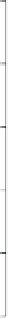Chemistry Reference
In-Depth Information
Energy
cutoff
9.0
1 nM
8.5
O
N
Under
predicted
N
O
8.0
N
7.5
N
Below
cutoff
N
7.0
True
Negative
6.5
-54
-52
-50
-48
-46
-44
-42
-40
-38
High
Score
Low
Score
E
inter
kcal/mol
Figure 9.12
Plot of observed activity versus energy score for compounds sampling the S1
pocket of BACE-1. (
) Compounds containing amine reagents which were incorporated prior
to the study; (
) compounds containing amine reagents selected independent of the scoring;
(
•
) compounds containing amine reagents selected based on scoring.
Figure 9.12). In addition, our arbitrary cut-off for the energy score may have missed some
interesting inhibitors, e.g. the ethyl- and isobutylamines which are labeled as 'Below cutoff'
in Figure 9.12. However, the score versus activity trend is clear, including a 'true negative'
control, the
tert
-butylamine reagent, which was predicted (and subsequently observed) to
yield an inhibitor with very low BACE-1 activity. In addition to demonstrating the utility
of an '
in situ
' virtual reagent selection, this study served to validate the hypothesis that the
BACE-1 S1
pocket is large, open and promiscuous, as a variety of reagents led to potent
BACE-1 inhibitors.
While BACE has been a favorite target for modelers as described above, the active site
of BACE has been discovered to have a high degree of flexibility, making virtual screening
particularly challenging. As seen in Figure 9.13, there are significant differences in several
loop conformations. It has been shown that the choice of P2/P3 substituents can affect the
the so-called 10s loop. Wild-type BACE exhibits 10s loop up, with an S10-T232 distance
>8 Å.
[
38
]
If the P3 substituent is large, then the 10s loop will remain up. If the P3 substituent
is small and the P2/P3 linker interacts with the T232 hydroxyl, then the 10s loop will remain
up and activity will be compromised. However, if the P3 substituent is small and the P2/P3
linker does not interact with the T232 hydroxyl, then the 10s loop will be down, creating a
more effective binding pocket for the small substituent.
[
39
]
Capturing this protein flexibility computationally is required to more accurately score
virtual molecules to prioritize synthesis of compounds. Figure 9.14 depicts the correlation
between binding energy calculated using MMFF
[
36
]
and measured pIC
50
for a diverse set of
ligands. The correlation is very high (
R
2
0.89) if the most appropriate 10s loop position
is chosen; however, if one uses only 10s up (down) the
R
2
=
is reduced to 0.32 (0.01).
[
33
]