Image Processing Reference
In-Depth Information
(b)
(a)
src
src
dst
dst
(c)
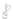
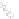
Fig. 22.1
Visualization methods for comparative genomics data. Most comparative genomics data
is shown using two kinds of encoding:
a
color encoding, where each chromosome in the destination
genome is assigned a unique color, and paired features in the source genome are colormapped
according to their paired feature's chromosome;
b
connection encoding, where lines are drawn
between the source and destination genomes connecting paired features.
c
MizBee [
28
] uses both
color and connection for showing conserved genomics data across multiple scales and in conjunction
with similarity scores
There are two predominate ways for visually representing the conserved pairs:
using color and using connection. To use color, as shown in Fig.
22.1
a, each chromo-
some in the destination genome is assigned a unique color. Each conserved feature
in the source genome is colormapped according to the chromosome its paired fea-
ture is associated with. Examples of visualization tools that use this encoding are
SyntenyVista [
14
], Sybil [
46
], and Cinteny [
42
]. Using color to encode conserved
features benefits from minimal visual clutter as the number of pairs increases, but
does so at the expense of only showing the location of the features on the source
genome. This encoding, however, suffers from color indistinguishability. We are able
to distinguish less than a dozen colors when showing categorical data [
50
], but most
genomes of interest will have on the order of dozens of chromosomes.
The second method for encoding conserved pairs is connection, shown in
Fig.
22.1
b. In this encoding, the source and destination genomes are aligned,













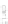














































































































Search WWH ::

Custom Search