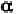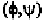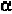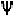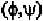Biology Reference
In-Depth Information
149
-149
0
,+69
0
-48
0
,-56
0
-61
0
,-35
0
-61
0
,-62
0
-49
0
,-52
0
-55
0
,-54
0
-48
0
,-59
0
20
21
22
23
24
25
26
Asp
Val
Ala
Gly
His
Gly
Gln
Residues 4 to 17 all have angles close to the -helical configuration.
Residue 18 is more close to the 3
10
-helical configuration (Dickerson and
Geis, 1969). Residue 20 is definitely not -helical. One may ask how
accurate these values are. This turns out to be a question not usually
answered by X-ray crystallographists. Instead, it is important to determine
the three-dimensional structure by a different method.
For horse cytochrome c consisting of 104 amino acid residues, its three-
dimensional structure has also been investigated by nuclear magnetic
resonance (Kar
et al
., 1994) measurements. There are about ten residues
having quite different angles from those determined by X-ray
diffraction studies. Such difference can be attributed to the different
physical states, one being in solution and the other in crystal. However,
they may also be due to the inability of positioning peptide units precisely
into the electron density map provided by X-ray diffraction analysis. To
connect the nearest -atoms with backbone atoms, it is possible to offset
the angle by 180
0
and the angle of the following residue also by 180
0
.
Hopefully, such errors are relatively rare.
Thus, for all proteins with known atomic coordinates of their non-hydrogen
atoms, the angles of each amino acid residue can be calculated, except
those at the N-terminal and C-terminal ends. They are associated with the
known amino acid sequences of these proteins. If indeed the sequence
determines the three-dimensional folding of a protein, these angles can
be closely correlated with the sequence. This valuable experimental
information unfortunately cannot be derived theoretically, without
extremely complicated calculations.
SELECTING AND ANGLES
In order to predict the three-dimensional folding of any protein with known