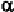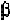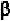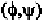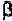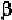Biology Reference
In-Depth Information
CHAPTER 6
PREDICTION OF PROTEIN FOLDING
INTRODUCTION
Since the classical experimental demonstration that the primary amino acid
sequence of a protein contains the information for three-dimensional folding
by Anfinsen (1973), numerous methods of prediction have been published in
the literature. As we have discussed in the previous Chapter, proline and
glycine residues can have important effects on the folding of the protein
backbone.
Due to the absence of the H-atom on the backbone of the proline residue, it
cannot form hydrogen-bonds in front of the N-terminal side of the protein
chain. Thus, it is usually considered to be an -helical breaking residue. In
some cases, its presence results in abend ofthe -helix. At the same time,
since its angle is around -60
0
which is somewhat different from that for
either anti-parallel or parallel -strand, proline is also considered as a
-strand breaker, or at least distorts a -strand.
On the other hand, glycine residue can have many angle combinations,
not possible for any other amino acid residue. Therefore, it has a tendency
of being present in bends or turns of protein backbones.
As a result, many of the protein folding predictive methods rely on such
statistical results of various amino acid residues being present in -helices,
-strands, or in turns, as suggested from three-dimensional structures of
proteins determined experimentally. Most of the methods are difficult to
understand. One of these (Chou and Fasman, 1974) was relatively straight
forward. However, sometimes one segment may have tendencies of being
-helical as well as forming -strands. Rules were then proposed to
choose one of these two possibilities. In general, most of the methods give