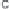Biology Reference
In-Depth Information
using Arithmetic averages), are more satisfying from a phylogenetic point
of view than others (WPGMA is not as highly sensitive to sampling bias
as UPGMA is).
4.2.1. The neighbor joining algorithm
An example of a more sophisticated and more phylogenetically valid
method is neighbour joining (NJ), which operates on a numerically
derived evolutionary distance. In contrast to raw dissimilarity, this
evolutionary distance tries to satisfy the property of additivity ([
AB
]
+
[
AC
]).
The NJ algorithm is based on the following four-point property:
given a tree with additive distances and four leaves
A
,
B
,
C
,
D
, of the
three possible sum of distance pairs
D
AB
[
BC
]
=
D
BD
, and
D
AD
+
D
BC
, two must be equal and the third must be smaller than the other two
(see Fig. 8).
NJ is a much-studied and widely accepted method that takes the het-
erogeneity of evolutionary rates in the different lineages into account,
but cannot detect homoplasy.
+
D
CD
,
D
AC
+
4.2.2. A common NTP artefact
Not only do NTP methods fail to allow for homoplasy, but most of them
also tend to draw together slowly evolving lineages, while moving quickly
evolving lineages to a basal (external or peripheral) position in the tree.
Figure 9 shows a hypothetical evolutionary tree (the true tree) and an
Fig. 8.
The NJ algorithm is based on the four-point property, which can be stated
as follows; for any four-leaf tree with additive distances, of the three possible sum of
distance pairs, two must be equal and the third must be smaller than the other two.