Information Technology Reference
In-Depth Information
2. Generate a phylogeny that represents the relationships between the se-
quences (the so-called
guide tree
) based on the scores of the pairwise
alignments.
3. Carry out the multiple alignment successively according to the guide tree,
starting with the sequences that are classified by the guide tree as most
similar.
The algorithm can furthermore directly generate phylogenetic trees for the
alignments, since it calculates the evolutionary distances between the se-
quences during the alignment computation.
ClustalW is actually only part of the Clustal program [182], which com-
prises ClustalW as a command line interface and ClustalX as a graphical user
interface. The software accepts a wide range of sequence formats as input, and
likewise supports a number of different output formats. It maintains default
settings for the main input parameters, so that users can directly start align-
ing sequences. In particular, however, it is often useful to change parameters
like the gap opening penalty and the gap extension penalty in order to obtain
better alignment results.
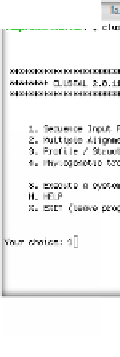
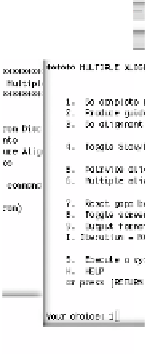
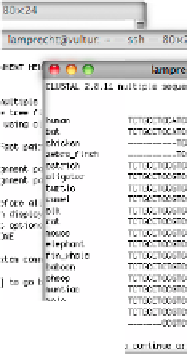
Fig. 3.5
ClustalW execution via the command line interface
Figure 3.5 gives an impression of the ClustalW command line interface:
After starting the program, the user is shown a list of functions to select
from. Usually the first thing to do in a ClustalW session is to load the input
sequences by selecting 1 and then entering the respective file name. Once the
sequences have been provided, the user is shown the list of functions again.
This time he can, for instance, choose 2 (Multiple Alignments) in order to
compute a multiple sequence alignment from the input sequences. ClustalW
prints elaborate status information during the algorithm execution, informing
about the current state of execution (pairwise alignments, guide tree, multiple





Search WWH ::

Custom Search