Information Technology Reference
In-Depth Information
in just two dimensions. Consequently, algorithms are used that follow reli-
able heuristic strategies. The popular ClustalW [320], for instance, computes
fast pairwise alignments of the input sequences in order to establish a so-
called guide-tree, which is then used to settle the order in which the multiple
alignment is successively assembled from the sequences (cf. Section 3.1.3).
Fig. 3.4
Multiple sequence alignment and derived phylogenetic tree
Sequence alignments define
distances
between sequences. Roughly speak-
ing, high sequence identity suggests that the sequences in question have a
comparatively young most recent common ancestor (i.e., a short distance),
while low identity suggests that the divergence is more ancient (a longer
distance). Figure 3.4 gives an example of a (part of a) multiple sequence
alignment and a derived phylogenetic tree. There are a number of distance-
based methods for the construction of phylogenetic trees, among the most
popular are the UPGMA algorithm [221] and the Neighbor-Joining method
[268]. A detailed elaboration on this topic would go beyond the scope of this
topic, for understanding the presented examples is it sucient to know that
multiple sequence alignments provide one possible basis for the estimation of
phylogenetic trees.
3.1.3 ClustalW
ClustalW [320] is the probably most popular multiple sequence alignment
program. The algorithm behind it utilizes the fact that similar sequences are
usually homologous [279, p. 81] and computes a multiple sequence alignment
in three major steps:
1. Compute pairwise alignments for all sequence pairs.

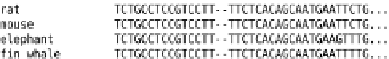










Search WWH ::

Custom Search