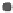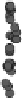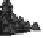Information Technology Reference
In-Depth Information
350
N
NSD
P
NSD
nfp
P
NSD
nfp
NSD
300
P
NSD
p
P
NSD
p
NSD
NSD
1
Trypsin-like
domain
(438)
250
200
150
NSD
0
100
50
Non-member
neighbors (1108)
TRYPSIN
0
0
100
200
300
400
500
NSD
C = TP
NSD
+ P
p
(NSD), + P
nfp
(NSD), + TP
AVS
+ P
p
(AVS) + P
nfp
(AVS),
Figure 1.
The principle of classifying domains in SBASE [14] (See text for
explanations).
Alignments can be sorted according to their position within the query, as well as
according to their common sequence patterns. Recent versions of BLAST, incorporate
position specific scoring known from profile methods (PSI-BLAST) as well as pattern-
specific searches (PHI-BLAST) [19-21].
Given the ease and speed of current sequence alignment algorithms, approximate
methods based on unstructured descriptions are used only in specific applications. In
composition-based methods, the sequences are described as vectors, in terms of the amino
acid, dipeptide, tripeptide etc. composition, and the comparison is based on simple
distances such as the Euclidean distance. Same as with other unstructured descriptors, the
calculation is very fast, especially since the database can be stored in the form of pre-
calculated vectors. The number of vector components (the resolution of the description) has
to be selected with care, and this is done either heuristically, or using an algorithm to
automatically select and/or weight those amino acid words that give the best separation
between a test group and a control group. In this manner group-specific distance functions
can be developed. The resolution of the description can be fine-tuned e.g. by decreasing the
amino acid alphabet (to 4,5, etc. letter alphabets instead of 20) and or by increasing the
word size (dipeptides, gapped dipeptides, tripeptides etc.). Examples include the
composition-based protein sequence search of Hobohm and Sander [22], as well as the
promoter-search program of Werner et al. [23-25]. Simple applications include the
recognition of coding regions based on codon-usage. Composition-based methods are very
useful for building recognizers for any sequence group for which a sufficient number of
examples are known. Given a test group and a control group of sequences, one can compare
the frequency of arbitrary words (provided as a list) between these two groups. The most
characteristic words can be selected based on simple measures such as the Mahalanobis
distance, and used for recognizing potential new members of the test group [26]. Similar
algorithms are often used in gene prediction systems [27].
Distributions are less frequently used for representing sequences, even though
methods of comparing sequence profiles such as hydrophobicity plots, secondary structure
propensity plots were developed already in the 1980-es. Fourier transforms of