Information Technology Reference
In-Depth Information
user can visualise the results of the analysis programs. PIX includes BLASTP searches
against NRL3D [33]. The transmembrane prediction programs PHDhtm [5], TopPred2
[34], MEMSAT2 [35] TMPred [36] and DAS [37] were used.
2.3. Generation of Multiple Sequence Alignments and Phylogenetic Analysis
The C-termini of forty SAND sequences (Tables 4a and 4b) were aligned in preparation for
phylogenetic analyses using the alignment program Clustalw (version 1.83) [38]. The N-
termini sequences were not included as they were too heterogeneous across the species.
Phylogenetic analysis was performed using PHYLO_WIN (version 1.2) [39]. SEAVIEW
was used to convert the alignment from MSF format to MASE format. PHYLO_WIN was
used to obtain a phylogenetic tree in ASCII format using the neighbour joining method,
with observed divergence, pairwise gap removal and 500 bootstrap replicates. The
character-based tree from PHYLO_WIN was rendered using the phylip drawtree program
(Figure 2). Based on this tree a subset of eleven of these sequences were chosen, as being
representative of distant taxa (Figure 3), and these were used for further protein sequence
analysis and structure prediction. The JEMBOSS Alignment Editor was used to view and
annotate sequence alignments (Figure 3) and to generate a percentage pairwise sequence
identity matrix (Table 5). JEMBOSS [40, 41] is the graphical interface to EMBOSS [42].
This
suite
of
programmes
is
freely
available
at
the
following
site
http://emboss.sourceforge.net/.
2.4. Secondary Structure, Solvent Accessibility and Fold Prediction
The secondary structure and solvent accessibility predictions were carried out using the
Jpred server [4,43]. The ClustalW alignment of the eleven representative SAND members
(Figure 3) was used as input to the Jpred server. The three fragments defined at the end of
Section 3.4 were analysed using the protein structure prediction MetaServer at
http://BioInfo.PL/meta [10]. This server submits the query-sequence to several servers that
perform structural fold predictions, the results are collated, summarised and consensus fold
predictions provided. SeqFold [44-45] and profiles-3D [45-46] were used to predict the
protein fold of the C-terminal section of SAND.
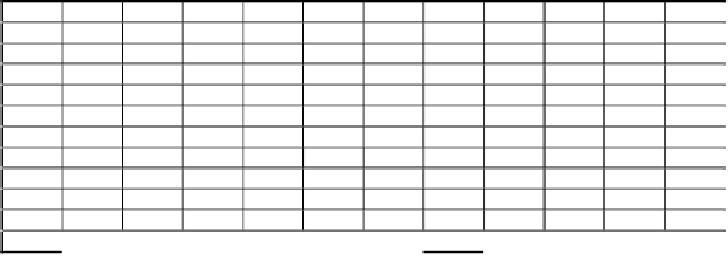
Table 5.
A matrix showing the pairwise percentage sequence identity of the SAND
proteins in Figure 3. The percentages are calculated using the JEMBOSS alignment
editor.
HS_1
FR_1
HS_2
FR_2
CI
DM
AT
OS
CE
PY
SP
HS_1
100.0
77.5
55.0
59.4
58.4
53.8
41.4
39.3
39.7
35.1
41.6
FR_1
100.0
53.6
56.7
58.4
51.9
42.0
40.3
41.4
37.2
42.2
HS_2
100.0
63.6
47.1
42.8
36.5
34.0
32.4
25.8
33.8
FR_2
100.0
49.6
43.9
35.5
33.4
33.6
29.4
33.6
CI
100.0
51.5
43.1
42.2
39.1
36.6
42.0
DM
100.0
38.4
37.8
40.5
35.1
37.2
AT
100.0
74.2
31.9
33.2
34.2
OS
100.0
31.3
34.0
34.5
CE
100.0
30.2
33.6
PY
100.0
33.8
SP
100.0